Philippines – Clinical Trial Regulatory Process

The Philippines is a country with a large population and a wide network of hospitals, medical centers, and healthcare professionals, making it a fertile platform for conducting clinical trials.

In clinical trials, the Philippines ranks number three in Southeast countries (US NIH) and ranks number eight worldwide (European Medicines Agency reports).
Clinical trial statistics in the Philippines
A total of 1,258 have been registered to date as per clinicaltrials.gov, with 111 ongoing studies. (Last update: 21/09/2024.
- Infectious Diseases: Including studies on tuberculosis, dengue, and COVID-19.
- Cancer: Oncology trials are significant due to rising cancer rates.
- Cardiovascular Diseases: Heart diseases are a leading health concern.
- Diabetes: With increasing prevalence, diabetes-related trials are common.
- Neurological Disorders: Research on conditions like stroke and dementia is also prominent.
The regulatory authority for clinical trials in the Philippines?
Clinical trials in the Philippines are regulated by the Department of Health (DOH) and the Food and Drug Administration (FDA). The FDA is an agency under the DOH and is responsible for ensuring the safety, efficacy, and quality of food, drugs, cosmetics, and medical devices.
Philippines clinical trial regulatory overview
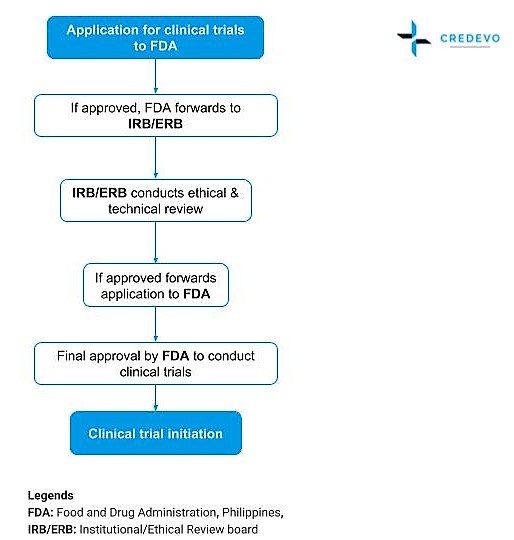
- To conduct clinical trials in the Philippines that involve the recruitment of Filipinos as volunteer subjects, the FDA (Food and Drug Administration, Philippines) approval is mandatory.
- Clinical trials can only be initiated after receiving approval from the Philippines FDA and Ethics Committee.
- The Center for Drug Regulation and Research (CDRR) in the Philippines is responsible for evaluating clinical trial proposals submitted to the FDA.
- FDA conforms to the highest ethical and technical standards of clinical research, The final approval by the FDA to conduct a trial will be based on a review Institutional Review Board/Ethical Review Board (IRB/ERB)
- All the establishments that will conduct clinical trials in the Philippines for the purpose of product registration, must obtain a Licence-To-Operate (LTO) from the FDA as a Clinical Trial Sponsor and/or Clinical Research Organization (CRO)
- All clinical trials shall be conducted in accordance with the latest International Council of Harmonization (ICH) and Good Clinical Practice (GCP) guidelines.
Review Board
- Applications approved by the FDA are forwarded to the Institutional/Ethical Review Board.
- ERB/IRB is accredited based on the recommendation of the Philippine Health Research Ethics Board (PHREB) by the FDA, Philippines.
- The timeline of IRB/ERB for decision-making on approval of the trial should not exceed 60 days.
- The FDA will give the final decision to approve or deny an application based on the recommendation, submitted in written format, emanating from the ERB/ERC review.
Application format
Documents are submitted as hardcopy or electronic files based on the preference of the FDA and ERB/ERC.
Documents submitted should include those in Parts A, B, and C, and such other documents or data as hereinafter be required by the FDA to ascertain the safety, efficacy, and quality of the products that will be subject to clinical study.
PART A
Clinical trial protocol and other pertinent documents
- Name and dosage form of product
- Title and aim of the trial
- Description of the trial design and subjects
- Treatment profile and operational aspects
- Adverse events and evaluation of results
- An informed consent form, case report form, and patient information sheet
- Resumes of principal and other investigators
- For multi-center studies, a list of principal investigators (and CVs) including trial sites
PART B
Pharmaceutical data
- GMP statement from manufacturing and certificate from a regulatory body
- Certificate of analysis and stability data.
- Manufacturing data, formulation, and product labeling.
PART C
Investigator’s Brochure IB (Efficacy and Safety Data)
Safety Data
- Non-Clinical Studies
- Clinical PK/PD and toxicology Studies, risks, and ADR anticipated
- Marketing experience, Periodic Safety Update Reports (PSUR), product status if marketed abroad
Efficacy data
- PK/PD data in human subjects
- In-house preliminary data
- Summaries of clinical trial studies conducted (Phase I, II, III)
- Published clinical data
What are the clinical practice guidelines for Ethical clearance?
In 2006, the Philippines government issued the National Ethical Guidelines for Health Research to reinforce clinical trial regulations and ensure that the regulatory phases of trials are followed.
The guidelines state seven criteria for ethical clearance of a research protocol:
- Science
- The nature and gravity of the risk to human subjects
- The adequacy of safeguards and protection against risk
- The magnitude of potential benefits or harm to individuals or the community
- The validity of the study participants’ informed consent
- The ecological impact
- Clarification of potential conflicts of interest [2]
Processing fee
- The initial submission fee is PHP 2500 (approximately US$50).
- Another PHP 20,000 to 40,000 (US$400 to US$800) is charged for IRB/ERB processing fees.
Approval process
- The applicants must submit their clinical trial proposals to the FDA as an electronic copy
- CDRR evaluates the documents and determines if
- The study has scientific value and is worth pursuing/conducting
- Conducted by duly licensed establishments
- Compliant with GCP, and other applicable best practices
- Compliance with the safety and efficacy standards of ICH
- The FDA passes these applications along to the Institutional Review Board/Ethical Review Board (IRB/ERB) for the conduct, of ethical and technical review.
- The IRB/ERB submits its recommendations for the approval or denial of the proposal to the FDA.
- The FDA makes the final decision to approve or to reject proposals.
Approval timeline
After submission,
- It’s about a 3-5 month process for clinical trial approval
- 60 calendar days for clinical trial amendments
- 30 days for the import permit
Clinical trial registry
All clinical trials should be uploaded to the Philippine Clinical Trial Registry (CTR) after 30 days from granting approval to conduct a clinical trial.
Clinical trial import license
- An import permit is granted after approval to conduct a clinical trial upon application supported by the FDA document attesting to the approval of the clinical trials to proceed based on compliance with ethical and technical requirements as ascertained by the ERB/ERC.
- The application may be submitted by the principal investigator/authorized representative/CRO with a permanent Philippine address, representing the sponsor through a letter of authorization.
- The import permit will be issued by the Product Services Division (PSD) with the cooperation of Regulation Division I, which has a linkage with the Bureau of Customs in this regard.
- The sponsor or CRO shall notify the FDA quarterly for every shipment of IPs and ancillary supplies entering the country.
Inspections
- FDA conducts random inspections on the clinical trial sites to monitor compliance with the approved study protocol and monitoring plan of the sponsor.
- It specifically looks into adherence to the GCP.
Safety reporting
- All adverse Drug Reactions (ADRs) both serious and unexpected are subject to expedited reporting.
- Fatal (deaths) or life-threatening, serious unexpected ADRs occurring in clinical trials, onsite, or offsite (for multi-site studies) should be reported. within 7 calendar days.
- All other unexpected serious ADRs, that are not fatal or life-threatening, onsite or offsite, must be filed as soon as possible, no later than 15 calendar days after first knowledge by the sponsor that the case meets the minimum criteria for expedited reporting.
- Adverse drug reactions that are not serious will also be reported in the regular progress report and final report. [3]
Termination of clinical trial
The CRO or Sponsor shall inform the FDA within 30 calendar days when the trial has been completed (the last investigational site has been closed) and or the trial has been closed globally.
Looking for regulatory support in the Philippines?
Credevo offers a wide range of drug development and regulatory services in the Philippines. Provide your query details below to connect with us.
To explore more Southeast Asian regions, follow the links below
- Clinical Trials in Indonesia
- Malaysia – Why and How to Start Your Clinical Trials?
- Conducting clinical trials in Vietnam – why and how to start?
- Singapore Clinical Trial Regulatory Process
- Thailand’s Clinical Trial Regulatory Scenario – Simplified (Part – 1, Part – 2).
2 thoughts on “Philippines – Clinical Trial Regulatory Process”
Comments are closed.