United States: Drug Approval Regulatory Process – II

This article is a sequence of the Regulatory & Drug Approval Process in the United States, Part-I, which describes the Investigational New Drug (IND) approval process, New Drug Approval (NDA), and Generic drug approval process. In this article, we will discuss the market approval process of biologics, biosimilars, orphan drugs, and various drug approval pathways.

Which is the regulatory authority for biologic drug approval?
The Center for Biologics Evaluation and Research (CBER) regulates the biologic drug approval.
CBER regulates cellular products, products composed of human, bacterial, or animal cells (such as pancreatic islet cells for transplantation) or physical parts of those cells (such as whole cells, cell fragments, or other components intended for use as preventative or therapeutic vaccines).
- Gene therapy products
- Vaccines
- Allergenic extracts
- Antitoxins, antivenins, and venoms
- Blood, blood components, plasma-derived products
In the United States, biological products are subject to a different premarket pathway. The biologic products shall have a license under a Biologics License Application (BLA) to prove that it is safe, pure, and potent.
Need support for your drug registration in the USA?
Credevo offers expertise in drug product registration, clinical trial regulations, and many more services in the USA. Complete the form below to connect with us!
Requirements for submission
The IND must contain
- Information from preclinical studies
- The product’s pharmacologic effects and mechanism of action and information on its ADME.
- Chemistry, Manufacturing, and Control (CMC) information and other several documents
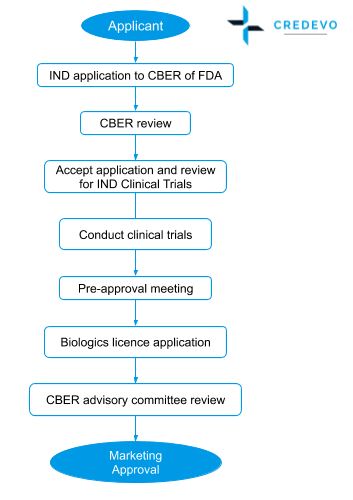
Biologics approval process
IND process
- Vaccines and biologics follow the same general pathway as for drugs.
- Sponsor shall submit all the non-clinical data in case of IND to FDA, FDA inaction in 30 days triggers the study under the IND to proceed
- The sponsor can initiate the clinical trials and process and review remains the same as of small molecules
- For every phase of a clinical trial, the sponsor has to review and continue to the next phase of the trial
- Once the clinical trial data is ready, the sponsor has to perform a pre-approval meeting.
NDA process
- After a sponsor submits a BLA, the FDA assembles a review team and then evaluates, within the first 60 days after submission, whether it can file the application or refuse
- For NDA the manufacturer shall submit an application to the Director, Center for Biologics Evaluation and Research (CBER) of FDA on forms prescribed for such purposes, and shall submit data derived from nonclinical laboratory and clinical studies to prove safety, purity, and potency
- Examination of biologics license application by USFDA:
- Examination
- Availability of product
- Manufacturing process
The new biological products receive 12 years of data protection
Process flow for the IND approval in the United States
Regulatory for biosimilars
Biosimilars are a type of biological product that is licensed by the FDA because they are highly similar to an already FDA-approved biological product and have been shown to have no clinically meaningful differences from the reference product.
All FDA-approved drugs, including biologics and their respective biosimilars, undergo a rigorous evaluation. Biosimilars and their biologic reference products each go through the specific regulatory approval process but there are differences. requirements for biosimilars.
The requirements for the biosimilars include
- Analytical studies
- Animal studies
- A clinical study or studies are sufficient to demonstrate the safety, purity, and potency of the proposed biosimilar product in one or more of the indications for which the reference product is licensed.
Along with the above data, interchangeable products shall include additional data
Labeling requirements
The labeling of a biosimilar product may differ from the reference product in many aspects, such as having fewer indications, differences in administration, preparation, storage, or safety information
Orphan drug regulatory
Designated orphan drugs and medical devices will be subject to a fast track drug development program, priority review, and accelerated approval.
The Center for Drug Evaluation and Research (CDER) reviews most orphan drug applications, and the Office of Orphan Products Development (OOPD) promotes orphan drug development and provides grants for the same. It is advised to contact these organizations regularly (Institute of Medicine (US) Committee on Accelerating Rare Diseases Research and Orphan Product Development; West).
Orphan Drug Designation
The Orphan Drug Act (ODA) grants special status to a drug or a biologic product that is intended to treat a rare condition or disease, upon the request of the sponsor. The status is referred to as Orphan designation or Orphan status
FDA provides several regulatory and financial incentives to develop the orphan drug in rare diseases
Before the sponsor can apply for drug approval under the Orphan Drug Act and before the sponsors are eligible for incentives like the Orphan Products Grant, they must apply and receive the orphan designation from the FDA (Institute of Medicine (US) Committee on Accelerating Rare Diseases Research and Orphan Product Development)
The US-FDA recommends applying before the IND studies, as it helps to design the trials when the requirements of clinical endpoints are known beforehand.
- The sponsor shall submit the designation request to the OOPD.
- The applicant does not need to have animal toxicology data. The sponsor requires only the results of efficacy studies in animal models for human disease (“Electronic Code of Federal Regulations”).
- The applicant may provide clinical data, animal studies, or in vitro data, but if sufficient information exists in published literature, that may suffice (Antos).
The nutraceuticals approval process in the USA
A dietary supplement is a product that contains one or more of the following dietary ingredients
- Vitamin,
- Mineral,
- Herb or other botanical,
- Amino acid,
- A dietary substance for use by humans to supplement the diet by increasing the total dietary intake of that ingredient; and
- A concentrate, metabolite, constituent, extract, or combination of any of the above.
FDA regulates dietary supplements under a different set of regulations than those covering conventional foods and drug products.
- For legal registration, the regulators classify the dietary ingredient into two categories.
- The product found in the market on or before 15 October 1994 is known as “Grandfathered” or “Old Dietary Ingredient” (ODI).
- The regulators allow these products to continue their stay in the market by checking their safety signals.
- The FDA considers the products placed in the market after 15 October 1994 or the changes made to the ODI as the New Dietary Ingredients (NDI).
- The manufacturer needs to get the premarket clearance from the FDA for the said products to place them in the US market.
- The Centre for Food Safety and Applied Nutrition (CFSAN) is the responsible body for the NDI.
Regulatory Process
- FDA does not approve the dietary supplement as any other drug product. It is the responsibility of the manufacturer/distributor to submit the relevant documents to claim the safety of the dietary supplement.
- The USFDA has released the GMP regulations for dietary supplements applied to all foreign companies and domestic companies.
- This GMP regulation specifies requirements for the Manufacture, Package, label, or hold of dietary supplements.
- Dietary supplements do not need approval from the FDA for marketing. Except in the case of a new dietary ingredient, where pre-market review for safety data and other information required by law.
When shall the manufacturer notify?
- The Dietary Supplement Health and Education Act (DSHEA) requires that a manufacturer or distributor notify FDA if it intends to market a dietary supplement in the U.S. that contains a “new dietary ingredient.”
- The manufacturer (and distributor) must demonstrate to FDA why the ingredient is reasonably expected to be safe for use in a dietary supplement unless it has been recognized as a food substance and is present in the food supply.
What are the various approval pathways?
Accelerated approval process
Accelerated drug approvals allow faster approval of drugs that treat serious diseases, the approval is faster because the effectiveness of drugs is based on the surrogate endpoints such as blood test, X-ray results rather than waiting for clinical trial results
Qualifying criteria
AND generally provides a meaningful advantage over available therapies AND demonstrates an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit or on an intermediate clinical endpoint
When could you apply?
Discuss early on development point
Features of this approval process
Approval based on the effect on the surrogate endpoint or intermediate clinical endpoint
Fastrack approvals
Here sponsors can submit portions of the application as the information becomes available (called rolling information) instead of waiting for all the data to come. This is for drugs for unmet needs or life-threatening diseases
Qualifying criteria
Would treat a serious condition (variously defined) AND nonclinical or clinical data demonstrate the potential to address an unmet medical need
When could you apply?
With IND application or open IND
Features of this approval process
Development and review are expedited, including the use of the rolling review
Breakthrough Therapy designation
Breakthrough Therapy designation expedites the development and review of drugs that are intended to treat a serious condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy. A drug with a Breakthrough Therapy designation is also eligible for the Fast Track process. The drug company must request a Breakthrough Therapy designation.
Qualifying criteria
AND preliminary clinical evidence indicates that the drug may demonstrate substantial improvement on a clinically significant endpoint(s) over available therapies
When could you apply?
With IND application or open IND
Features of this approval process
- All fast track features
- Intensive guidance on efficient drug development during IND
- Organizational commitment involving senior managers
Priority review
Priority review is a program of the United States Food and Drug Administration (FDA) to expedite the review process for drugs that are expected to have a particularly great impact on the treatment of a disease.
Qualifying criteria
AND if approved, would provide a significant improvement in safety or effectiveness
Features of this approval process
Shorter clock for review of marketing application (6 months compared to the 10- month standard review)
Do you need support or have queries on drug registration requirements in the USA?
Credevo offers a wide range of drug development and regulatory services in the USA. Provide your requirement details below to connect with us and explore our services.
References
- https://www.fda.gov/about-fda/center-biologics-evaluation-and-research-cber/center-biologics-evaluation-and-research-cber-responsibilities-questions-and-answers
- https://d2cax41o7ahm5l.cloudfront.net/cs/speaker-pdfs/shashi-kumar-yadav-sri-indu-institute-of-pharmacy-india.pdf
- https://www.fda.gov/files/drugs/published/Biosimilar-Product-Regulatory-Review-and-Approval.pdf
- https://www.tevabiosimilars.com/globalassets/biosimilars/videos-and-resources/Biosimilars_101_Brochure.pdf
- https://fas.org/sgp/crs/misc/R41983.pdf