Clinical Trial Start-Up Guide in Asia-Pacific: Regulatory Timelines, Ethics Approval (EC) Processes & IMP Import Requirements
Key takeaways
- Singapore and Malaysia lead APAC clinical trial start-up timelines with faster regulatory approvals, while Vietnam and Indonesia offer larger patient recruitment opportunities with higher operational complexity.
- Ethics Committee approvals, IMP import permits, translation requirements, and site contract negotiations are the most common causes of clinical trial delays across ASEAN countries.
- Parallel EC and regulatory submissions can significantly reduce APAC trial activation timelines, but approval processes remain fragmented and country-specific.
- Thailand and the Philippines are strong choices for Phase II and III clinical trials due to recruitment potential and established investigator networks.
- Successful APAC clinical trial execution depends on realistic feasibility planning, local CRO expertise, and early alignment of regulatory, EC, and import pathways.
Asia-Pacific (APAC) has become one of the most attractive regions for clinical trials due to patient diversity, strong investigator networks, and lower operational costs. However, clinical trial start-up timelines remain highly fragmented across countries.

Singapore remains the fastest and most predictable market for clinical trial activation due to centralized systems, parallel submissions, and efficient Health Sciences Authority (HSA) reviews. Malaysia follows with relatively stable timelines and improving NPRA responsiveness.
Thailand and the Philippines offer strong patient recruitment potential but often face EC variability and import permit delays. Vietnam and Indonesia can deliver high recruitment value but require careful regulatory planning due to layered approvals, translation requirements, and administrative complexity.
In practice, the “fastest” country is not always the most suitable one for every protocol. Regulatory predictability, ethics committee structure, import logistics, site readiness, and local operational complexity all influence overall study start-up performance. In some cases, a country with slightly longer approval timelines may still be strategically preferable because of stronger patient access, higher investigator engagement, or better long-term recruitment potential.
High-level APAC comparison
The comparison below provides a high-level overview of how these factors typically differ across major APAC clinical trial markets.
| Country | Regulatory Predictability | Typical Start-Up Speed | EC Structure | IMP Import Complexity |
|---|---|---|---|---|
| Singapore | Very High | Fastest | Centralized/Institutional | Low |
| Malaysia | High | Fast | Institutional + MREC | Moderate |
| Thailand | Moderate | Medium | Institutional ECs | Moderate–High |
| Vietnam | Variable | Slow–Medium | Multiple EC layers | High |
| Indonesia | Variable | Slow | Multi-layered | High |
| Philippines | Moderate | Medium | PHREB + Site ECs | Moderate |
APAC regulatory landscape overview
APAC has become a major destination for global clinical trials due to its large patient populations, expanding research infrastructure, and increasing regulatory maturity across many ASEAN markets. The region offers significant advantages for sponsors, including strong recruitment potential, access to diverse and treatment-naïve populations, and cost-efficient trial execution.
At the same time, APAC’s strength lies in the individuality of its markets. Each country operates through its own regulatory framework, ethics review process, and import pathway, allowing sponsors to select countries strategically based on study requirements, timelines, therapeutic area needs, and operational priorities. This country-specific approach makes APAC a highly adaptable region for clinical development across a wide range of trial designs and sponsor objectives.
How does clinical trial start-up work in APAC?
APAC clinical trial start-up involves working across a diverse group of regulatory systems, with each country offering its own approval pathways, ethics committee processes, import requirements, and documentation frameworks. This allows sponsors to strategically select markets based on the specific needs of their study, patient population, and development timelines.
Common themes across APAC
While APAC regulatory systems differ significantly from one country to another, several recurring operational and regulatory patterns consistently influence how clinical trial approvals, study start-up timelines, and submission strategies are managed across the region.
- Some countries allow parallel EC and regulatory submissions, enabling sponsors to streamline start-up timelines by submitting to both the regulatory authority and ethics committee simultaneously, while others follow a more structured sequential review process.
- Ministries of Health often play an important role in shaping and supporting clinical trial approval pathways within their respective countries.
- IMP import permits are typically integrated into the broader regulatory approval process to help ensure compliance and product oversight.
- Local legal representatives or CRO partnerships are commonly required, helping sponsors navigate country-specific operational and regulatory expectations more efficiently.
- Oncology, biologics, gene therapy, and rare disease studies generally undergo more comprehensive review processes due to their scientific and regulatory complexity.
Standard clinical trial start-up flow
A standard clinical trial start-up flow in the Asia-Pacific region typically involves a series of regulatory, ethical, and operational steps. However, these steps may vary by country; generally, they follow the sequence outlined below.
- Regulatory submission (CTA/IND equivalent): Submission of the clinical trial application to the national regulatory authority to seek approval to conduct the study.
- Regulatory authority & Ethics Committee review: Parallel or sequential review of the clinical trial application by both the competent regulatory authority and the ethics committee (central or institutional), assessing safety, scientific validity, and ethical compliance.
- EC approval (Central or Institutional): Formal approval from either a central ethics committee or an institutional ethics committee, confirming that the study meets ethical requirements and ensures protection of trial participants.
- IMP import license approval: Authorization to import investigational medicinal products into the country in compliance with local regulatory requirements.
- Site activation: Completion of regulatory, contractual, and operational start-up activities enabling the clinical site to initiate trial operations.
- First Patient In (FPI): Enrollment and dosing of the first eligible participant, marking the official start of clinical trial execution.
The difference lies in whether these activities occur in parallel or sequentially.
Country-by-country clinical trial start-up analysis
Now, let’s take a closer look at the country-by-country regulatory landscape in detail.
Clinical trial regulatory process – Singapore
Singapore has established itself as one of Asia’s leading clinical research hubs, particularly for early-phase and innovative therapies. Healthcare stakeholders widely recognize the country for its efficient healthcare system, strong regulatory oversight, and highly developed research infrastructure.
Regulatory authority in Singapore
In Singapore, clinical trials are regulated by the Health Sciences Authority (HSA), which oversees the approval of pharmaceutical, biologic, and medical device studies. Ethics review is conducted separately through IRBs or DSRBs. Singapore is known for its efficient and predictable clinical trial regulatory system within APAC.
Submission pathway
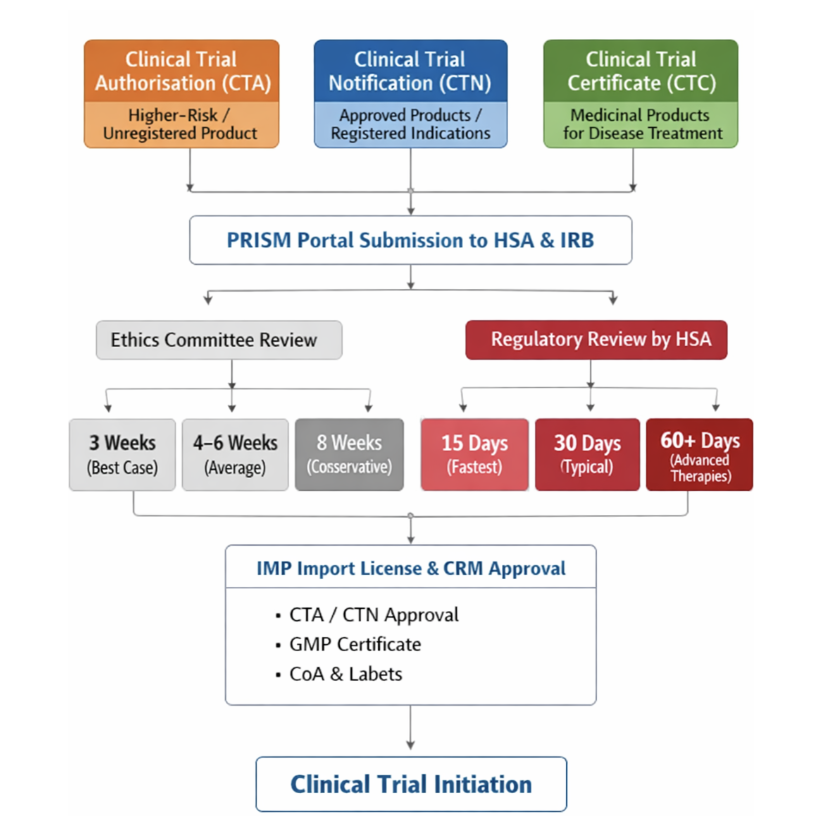
In Singapore, clinical trials are regulated through three main pathways, depending on the nature of the investigational product and its intended use
- Clinical Trial Authorisation (CTA): Required when the investigational product is not registered in Singapore or when the trial involves higher-risk studies that require full regulatory review before initiation. Approval must be obtained before starting the trial.
- Clinical Trial Notification (CTN): Used for clinical trials involving investigational products that are already approved in Singapore and are being used within their registered indication. These studies generally follow a simplified notification-based process.
- Clinical Trial Certificate (CTC): Required before initiating any clinical trial involving medicinal products for the treatment or prevention of diseases. This certificate confirms that the trial has met all regulatory requirements and is authorized to proceed in Singapore.
Clinical Research Material (CRM) refers to any investigational product or material used specifically for clinical research purposes in Singapore. When a CRM notification is required, it should be included within the same CTA, CTN, or CTC application. Sponsors can submit applications to both HSA and the IRB in parallel. All submissions must be completed through the PRISM portal.
Ethics Committee process
Primarily institutional IRBs with centralised operational efficiency.
Clinical trial approval timelines and key considerations in Singapore
The table below summarizes typical clinical trial approval, ethics, and import-related timelines along with common operational bottlenecks in Singapore.
| Category | Timelines / Requirements |
|---|---|
| EC Approval Timelines | Best case: 3 weeks Average: 4–6 weeks Conservative: 8 weeks |
| Regulatory Review Timelines | Fastest: 15 working days (certain Phase I studies) Typical: 30 working days Delayed: 60+ days (advanced therapies) |
| IMP Import License & Logistics | Managed under the integrated CTN/CTA process by the Health Sciences Authority Required documents: – CTA approval, – GMP certificate, – CoA, – label documentation |
| Common Bottlenecks | – Advanced therapy reviews – GMO-related assessments – Sponsor underestimation of CMC expectations |
Regulatory process flow in Singapore
Practical insights
- Singapore is one of the most operationally predictable clinical trial markets in APAC, with clear, transparent, and consistently applied regulatory processes that support reliable planning of study timelines.
- Compared to neighboring countries, CRO selection has less impact on start-up speed due to highly standardized systems and strong regulatory alignment.
- Additional strengths include efficient review timelines, high-quality research sites, and a well-established clinical research ecosystem, making Singapore particularly suitable for early-phase and complex studies.
While Singapore is often selected for speed and regulatory predictability, many sponsors eventually look toward Malaysia when seeking a balance between operational efficiency, regional access, and cost-effectiveness.
Malaysia has steadily strengthened its position as an attractive ASEAN clinical trial destination for mid- to late-phase studies. Let’s walk through Malaysian Regulatory Timelines
Clinical trial regulatory process – Malaysia
Malaysia has become increasingly attractive for regional and multinational studies due to its balance between regulatory efficiency and operational costs. The country offers experienced investigators, growing site capabilities, and relatively stable approval timelines.
Regulatory authority
In Malaysia, clinical trials are regulated by the National Pharmaceutical Regulatory Agency (NPRA), while ethics review is handled by MRECs or institutional ethics committees. It is generally a structured and predictable regulatory environment in APAC.
Submission pathway
Parallel EC and regulatory submissions are generally possible.
Two regulatory processes exist for clinical trials in Malaysia
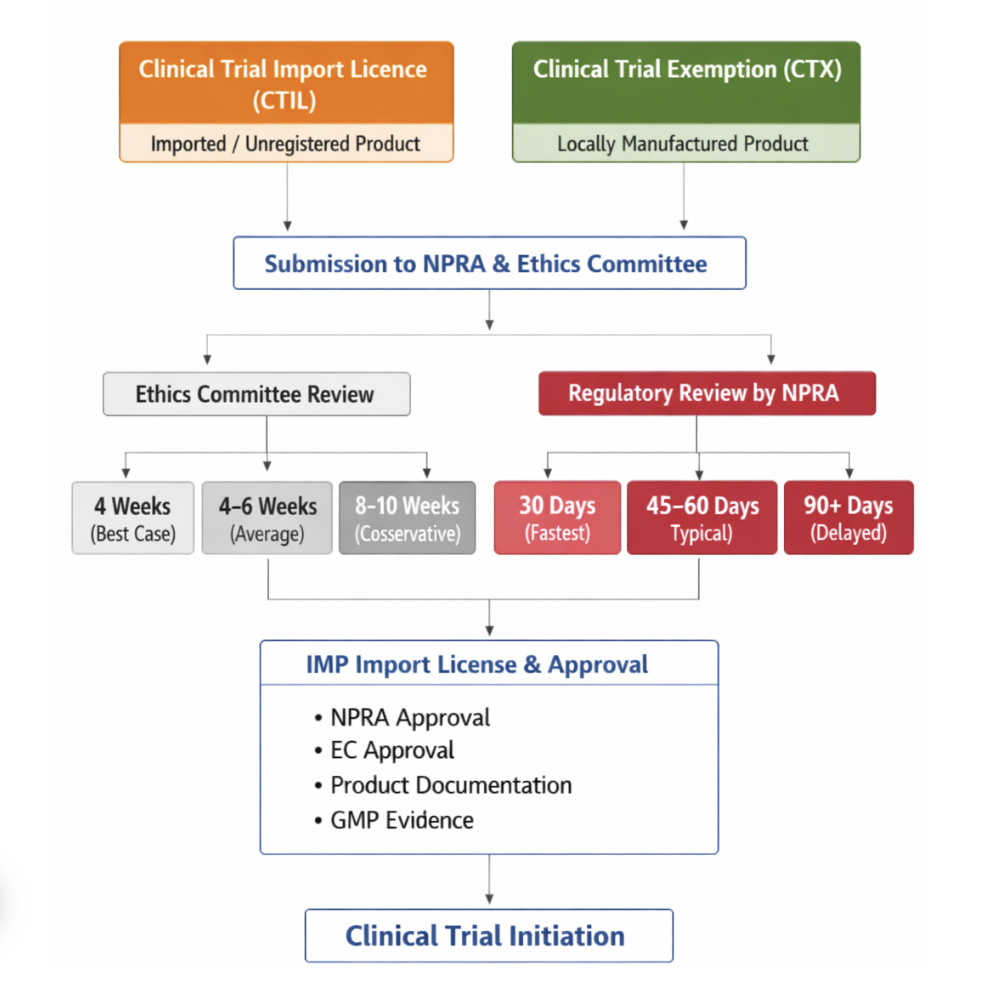
- CTIL Clinical Trial Import Licence
- CTX Clinical Trial Exemption
Clinical Trial Import Licence (CTIL)
A Clinical Trial Import Licence (CTIL) is required when the investigational product (IP) is manufactured outside Malaysia and needs to be imported into Malaysia for clinical trial use. It authorizes the sponsor or license holder to import an unregistered product specifically for the approved clinical trial.
Apply CTIL when:
- The IMP is manufactured outside Malaysia
- The product is unregistered in Malaysia
- You need to import a finished investigational medicinal product for use in the study
Clinical Trial Exemption (CTX)
A Clinical Trial Exemption (CTX) applies when a company manufactures the investigational product locally in Malaysia solely for clinical trial purposes, and NPRA grants an exemption from the standard product registration requirement.
Ethics Committee Process
Medical Research and Ethics Committee (MREC) or institutional ECs.
Clinical trial approval timelines and key considerations in Malaysia
The table below summarizes typical clinical trial approval, ethics, and import-related timelines along with common operational bottlenecks in Malaysia.
| Category | Timelines / Requirements |
|---|---|
| EC Approval Timelines | Best case: 4 weeks Average: 4–6 weeks Conservative: 8–10 weeks |
| Regulatory Review Timelines | Fastest: 30 days, Typical: 45–60 days, Delayed: 90+ days |
| IMP Import License & Logistics | Requirements: – NPRA approval, – EC approval, – product documentation, and – GMP evidence. Typical timeline: 1–3 weeks |
| Common Bottlenecks | – Site contracting – Budget negotiations – Delayed insurance documentation |
Regulatory process flow in Malaysia
Practical Insights
Malaysia offers one of the best balances between speed, cost, and regulatory predictability in ASEAN.
Now, let’s see how Thailand frequently enters the discussion due to its strong hospital infrastructure and experienced investigator networks. The country has become particularly attractive for oncology and recruitment-intensive clinical trials across Southeast Asia
Clinical trial regulatory process – Thailand
Thailand continues to attract global sponsors because of its strong hospital networks, experienced investigators, and high recruitment potential, especially in oncology and specialty therapeutic areas.
Regulatory authority
In Thailand, clinical trials are regulated by the Thai Food and Drug Administration (Thai FDA), with ethics review conducted by institutional ethics committees. Thailand is a well-established APAC clinical trial hub, though timelines can vary depending on review workload and documentation quality.
Submission pathway
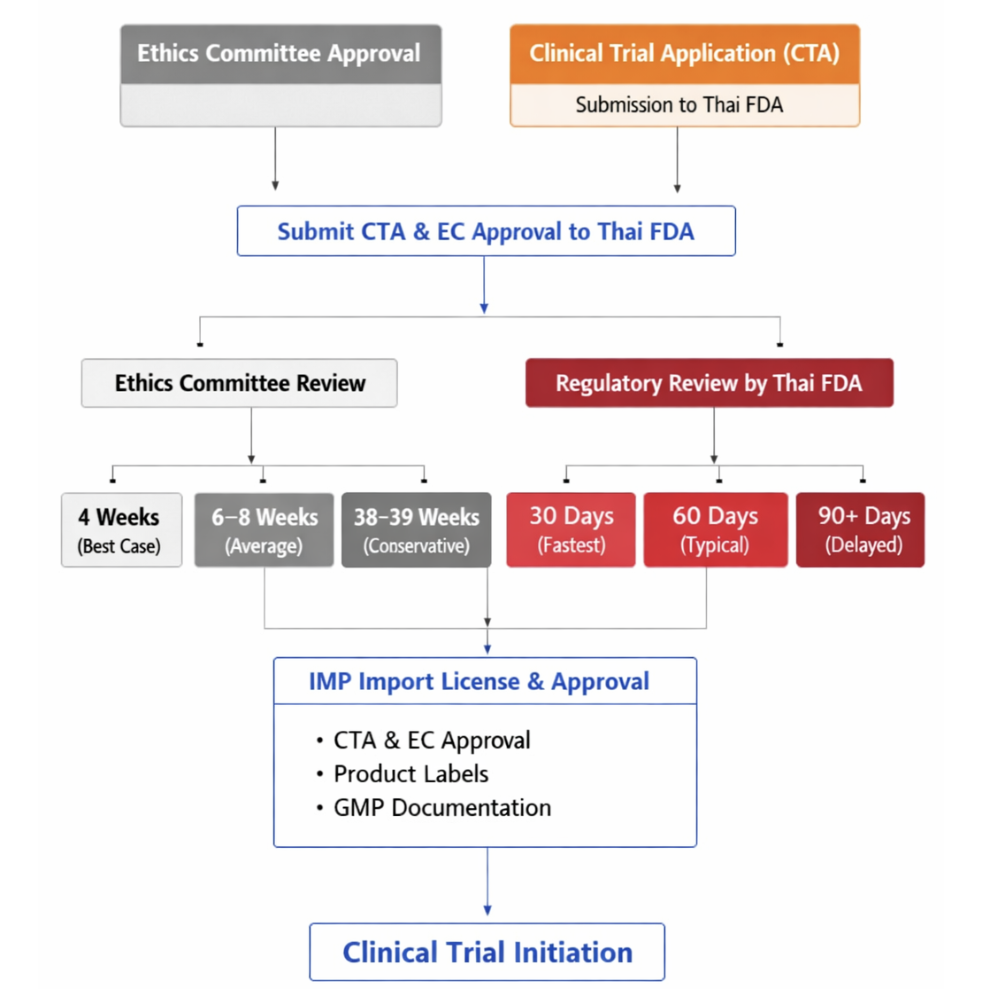
Sponsors submit a Clinical Trial Application (CTA) to the Thai FDA. The FDA-recognised Ethics Committee issues the EC approval required to conduct the clinical trial. After receiving EC approval, sponsors submit the CTA to the Thai FDA along with the EC approval letter.
Ethics Committee process
Institutional ethics committees (ECs) and other types of ECs, including the Ethical Review Committee for Research in Human Subjects, Ministry of Public Health (ECMOPH), and the Central Research Ethics Committee (CREC), should be recognized by the Thai Food and Drug Administration (Thai FDA) to review and approve clinical trial protocols.
Clinical trial approval timelines and key considerations in Thailand
The following table summarizes typical clinical trial approval timelines and key operational considerations across regulatory and ethics processes in Thailand.
| Category | Timelines / Requirements |
|---|---|
| EC Approval Timelines | Best case: 4 weeks Average: 6–8 weeks Conservative: 38–39 weeks |
| Regulatory Review Timelines | Fastest: 30 days Typical: 60 days Delayed: 90+ days |
| IMP Import License & Logistics | Issued by the Thai FDA after regulatory clearance. Required documents: – CTA approval, – EC approval, – product labels, – GMP documentation Typical import review: 2–4 weeks |
| Common Bottlenecks | – Multiple EC revision rounds – Thai language translation requirements – Contract negotiation delays |
Regulatory process flow in Thailand
Practical insights
- Strong clinical research ecosystem, particularly in oncology and specialty studies, with experienced investigators and good patient recruitment potential.
- Well-established regulatory framework, though institutional EC timelines may vary depending on the site.
- Site selection plays a key role in ensuring a smooth and efficient study start-up.
- Effective CRO and site collaboration can help improve consistency and operational efficiency in trial execution.
Moving into Vietnam introduces a very different regulatory environment. While Vietnam offers significant patient recruitment potential, sponsors often encounter more layered approval processes and greater operational variability compared to other ASEAN markets.
Clinical trial regulatory process – Vietnam
Researchers and sponsors increasingly recognize Vietnam for its large patient population and growing clinical research activity. However, sponsors often encounter more administrative complexity compared to neighboring ASEAN countries.
Regulatory authority
In Vietnam, clinical trials are regulated by the Drug Administration of Vietnam (DAV), with ethics review conducted by institutional ethics committees. Vietnam’s regulatory process is structured but can involve multiple review layers, leading to variable timelines depending on study complexity and documentation quality.
Submission pathway
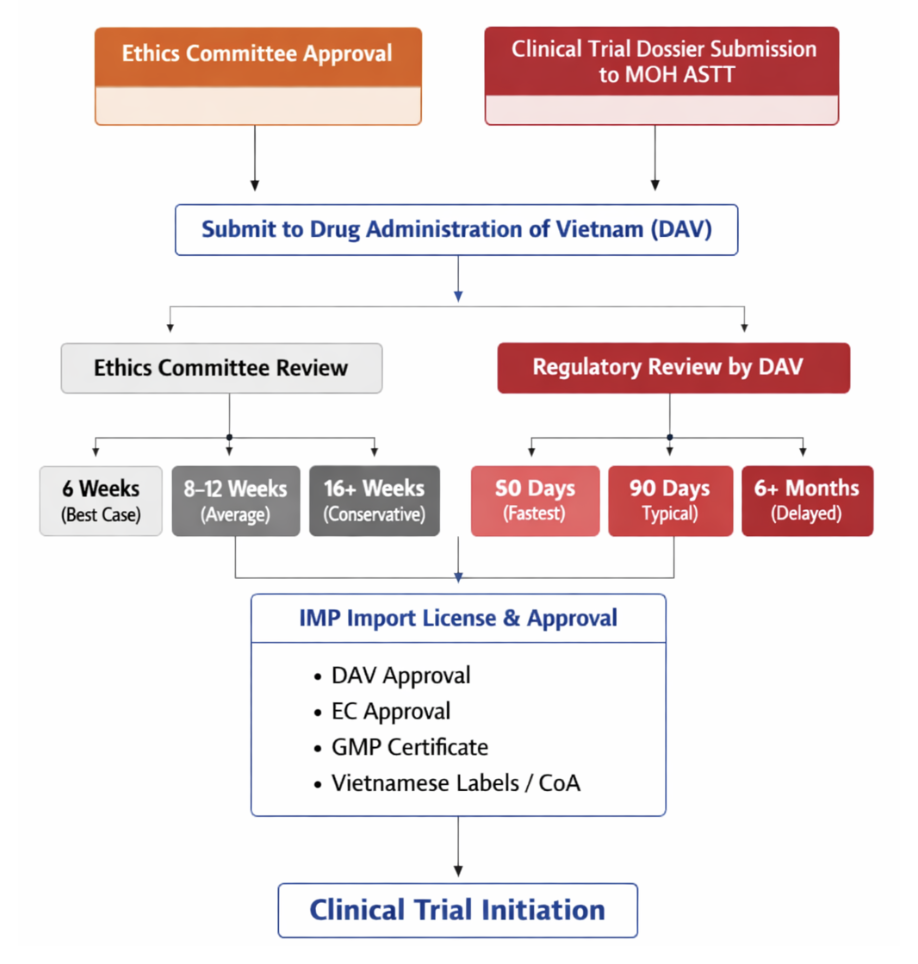
Sequential processes are common, especially for imported IMPs.Need a local CRO or PI to submit the application to MOH ASTT
The sponsor must submit, directly or via post, one (1) copy of the clinical trial registration dossier signed by the sponsor’s representative to the ASTT.
Ethics Committee process
Central and institutional EC involvement may both apply.
Clinical trial approval timelines and key considerations in Vietnam
The following table outlines typical clinical trial regulatory, ethics, and import-related timelines and key operational considerations in Vietnam.
| Category | Timelines / Requirements |
|---|---|
| EC Approval Timelines | Best case: 6 weeks Average: 8–12 weeks Conservative: 16+ weeks |
| Regulatory Review Timelines | Fastest: 60 days Typical: 90 days Delayed: 6+ months |
| IMP Import License & Logistics | Import permits are tightly linked to Ministry of Health approvals Required documents: – Regulatory approval, – EC approval, – GMP certificate, – CoA, – Vietnamese labels/translations |
| Common Bottlenecks | – Translation requirements – Administrative review cycles – Import permit sequencing |
Regulatory process flow in Vietnam
Practical insights
- Vietnam is an emerging clinical trial market with strong growth potential and increasing interest from global sponsors.
- Regulators structure the processes, but sponsors must manage approvals that are document-intensive and rely on early planning and local expertise.
- Site capabilities are improving, with growing experience in multinational trials, particularly in infectious diseases and general medicine.
- Site initiation timelines can vary, making proactive coordination with local partners important for a smooth start-up.
- With increasing regulatory maturity, Vietnam is becoming a more attractive destination for cost-efficient patient recruitment in APAC.
Similar to Vietnam, Indonesia presents an enormous long-term opportunity because of its large and diverse population. Yet, clinical trial start-up in Indonesia often depends heavily on local operational expertise, regulatory navigation, and import coordination.
Clinical trial regulatory process – Indonesia
Indonesia presents significant long-term potential for clinical research due to its large and diverse patient population. However, regulatory navigation and operational coordination remain key challenges for sponsors entering the market.
Regulatory authority
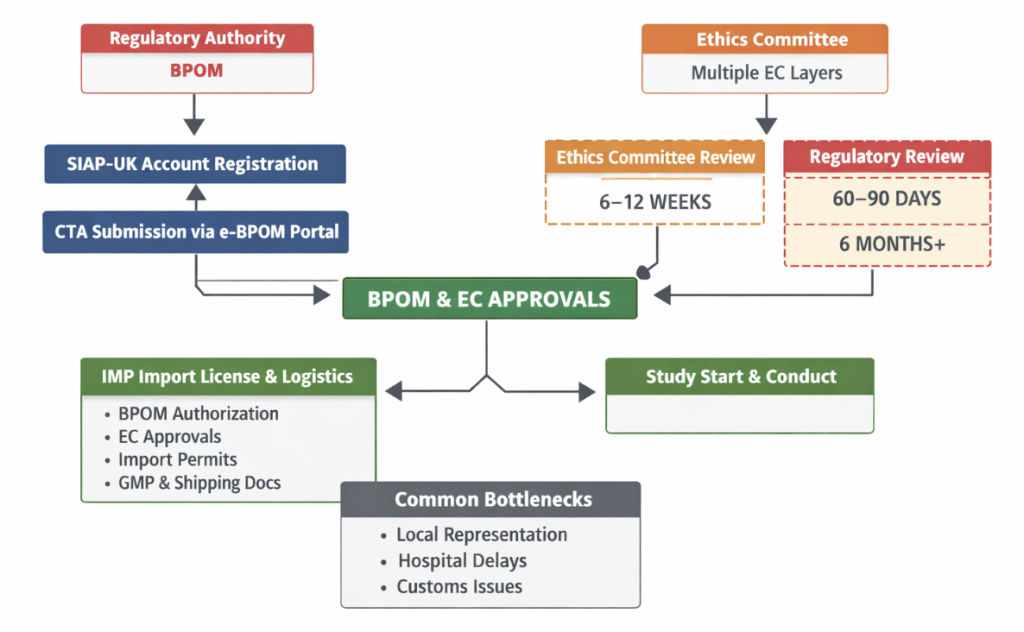
In Indonesia, clinical trials are regulated by the Indonesian Food and Drug Authority (BPOM), with ethics review conducted by institutional ethics committees. The regulatory process is multi-layered and can be complex, often resulting in longer and more variable approval timelines.
Submission pathway
Regulatory review often runs alongside local ethics approvals, but operational sequencing differs by institution. Regulatory framework established by the National Agency of Drug and Food Control (BPOM). So, A well-structured Clinical Trial Application (CTA) is crucial for obtaining approval from the BPOM Local Representative, and local CRO need to register on the BPOM online portal. SIAP-UK needs to create a company account and then submit the CTA via SIAP-UK. Then the CTA Application must be submitted through the e-BPOM portal e-bpom.pom.go.id
Ethics Committee process
Multiple EC layers may apply, especially for public hospitals.
Clinical trial approval timelines and key considerations in Indonesia
The table below summarizes typical regulatory, ethics, import, and operational timelines along with key bottlenecks for clinical trial startup in Indonesia.
| Category | Timelines / Requirements |
|---|---|
| EC Approval Timelines | Best case: 6 weeks Average: 8–12 weeks Conservative: 16+ weeks |
| Regulatory Review Timelines | Fastest: 60 days Typical: 90 days Delayed: 6+ months |
| IMP Import License & Logistics | Import permits require: – BPOM authorization, – EC approvals, – local importer documentation, – GMP and shipping records Typical import timeline: 3–6 weeks |
| Common Bottlenecks | – Local representation requirements – Decentralized hospital processes – Customs clearance delays |
Regulatory process flow in Indonesia
Practical insights
- Indonesia offers one of the largest patient pools in APAC, making it highly attractive for recruitment-heavy studies.
- Careful coordination is required to manage structured regulatory processes, approvals, and document-intensive import procedures.
- Site readiness varies significantly, so selecting experienced sites is key to avoiding start-up delays.
- Local partnerships are essential to navigate operational complexity and ensure smooth trial execution.
- Best suited for studies where patient access is a primary priority over rapid start-up timelines.
The Philippines, meanwhile, offers a more balanced clinical research environment within ASEAN. With experienced investigators and growing participation in multinational trials, the country continues to attract sponsors looking for strong recruitment potential alongside moderate regulatory complexity.
Clinical trial regulatory process – Philippines
The Philippines remains an important ASEAN market for multinational Phase III studies because of its experienced investigators and strong patient recruitment capabilities.
Regulatory authority
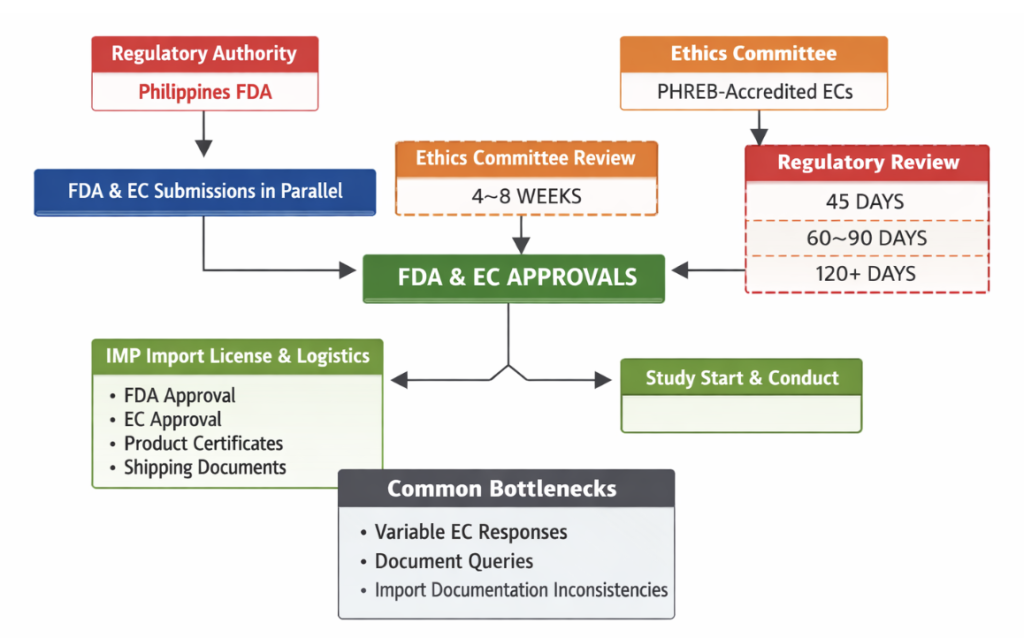
In the Philippines, clinical trials are regulated by the Food and Drug Administration of the Philippines (Philippine FDA), while ethics review is conducted by the Philippine Health Research Ethics Board (PHREB) and institutional ethics committees. The system is structured but may involve multiple review steps, leading to moderate variability in approval timelines.
Submission pathway
FDA and EC submissions may occur in parallel.
Ethics Committee process
PHREB-accredited ECs with institutional reviews.
Clinical trial approval timelines and key considerations in the Philippines
The table below summarizes typical clinical trial approval timelines, import requirements, and common operational bottlenecks in the Philippines.
| Category | Timelines / Requirements |
|---|---|
| EC Approval Timelines | Best case: 4 weeks Average: 6–8 weeks Conservative: 12 weeks |
| Regulatory Review Timelines | Fastest: 45 days Typical: 60–90 days Delayed: 120+ days |
| IMP Import License & Logistics | Import licenses require: – FDA approval, – EC approval, – product certificates, – shipping documentation Typical import timeline: 3–5 weeks |
| Common Bottlenecks | – Variable EC responsiveness – Administrative document queries – Import documentation inconsistencies |
Regulatory process flow in the Philippines
Practical insights
- The Philippines has a growing clinical research ecosystem with strong English proficiency and experienced investigators.
- Regulatory and ethics processes are generally clear, but timelines can vary depending on institutional review boards.
- Good potential for patient recruitment, especially in urban tertiary care hospitals.
- Import and logistics processes require close coordination with local regulatory and customs stakeholders.
- Overall, a balanced APAC market offering a mix of accessibility, cost efficiency, and operational familiarity.
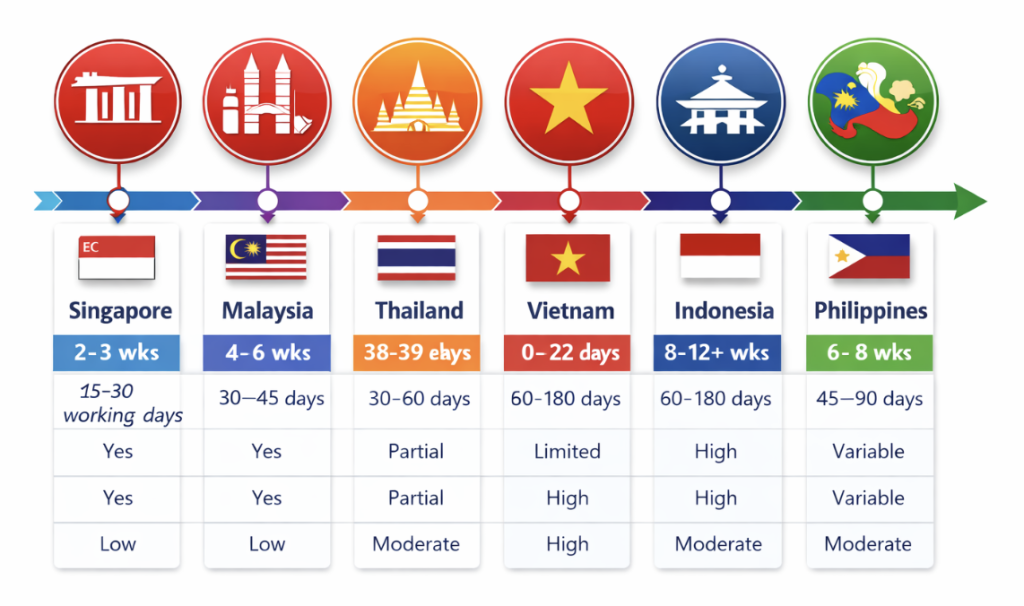
Comparative table: APAC trial start-up timelines
| Country | EC Timeline | Regulatory Timeline | Parallel Submission | Import Complexity | Predictability |
| Singapore | 2-3 wks | 15–30 working days | Yes | Low | Very High |
| Malaysia | 4–6 wks | 30–45 days | Yes | Moderate | High |
| Thailand | 38–39 wks | 30–60 days | Partial | Moderate | Moderate |
| Vietnam | 8–12 wks | 60–180 days | Limited | High | Variable |
| Indonesia | 8–12+ wks | 60–180 days | Partial | High | Variable |
| Philippines | 6–8 wks | 45–90 days | Yes | Moderate | Moderate |
APAC clinical startup timelines comparison
Strategic comparison
Fastest country for trial start-up: Singapore consistently delivers the shortest and most predictable clinical trial activation timelines in the APAC region due to its streamlined and well-standardized regulatory processes.
Most complex regulatory environment: Indonesia and Vietnam present relatively higher operational complexity due to multi-layered approvals, decentralized processes, and import permit dependencies.
First-in-human studies: Singapore is the preferred hub for FIH studies owing to its strong regulatory maturity, high-quality clinical infrastructure, and robust GCP-compliant research environment.
Phase II/III recruitment: Thailand, Vietnam, Indonesia, and the Philippines offer strong patient access and larger treatment-naïve populations, supporting efficient recruitment for mid-to-late phase trials.
Best for oncology trials: Singapore and Thailand continue to demonstrate strong oncology expertise, supported by experienced investigators and established clinical research networks.
Note: Please note that these insights are based on our previous experience and may evolve over time. We strongly recommend connecting with our team by filling out the form below to obtain study-specific regulatory processes, timelines, and country suitability assessments relevant to your particular clinical trial.
Common sponsor mistakes in APAC
- Assuming timelines are uniform across ASEAN
- Ignoring EC variability between hospitals
- Underestimating import permit timelines
- Selecting countries based only on recruitment cost
- Failing to align feasibility with regulatory reality
- Using inexperienced local vendors
Practical APAC trial start-up checklist
Before selecting APAC countries, confirm the following key elements are clearly understood and validated:
- EC pathway understood: including whether central, institutional, or hybrid ethics review applies in each country
- Regulatory sequencing mapped: clear understanding of submission order, dependencies, and approval flow
- Import permit process validated: requirements, timelines, and responsible parties for IMP importation confirmed
- Translation requirements identified: all essential documents and submission materials translated as per local regulations
- Site activation assumptions are realistic: timelines aligned with actual site readiness and operational capacity
- Parallel submission opportunities confirmed: possibility of concurrent EC and regulatory submissions verified
- CRO/local agent capabilities assessed: operational strength, regulatory experience, and site management capability evaluated
Conclusion
APAC remains one of the highest-growth regions for global clinical research, but successful execution depends on understanding structural differences between countries.
Singapore and Malaysia offer speed and predictability, while Vietnam and Indonesia provide scale with higher operational complexity. Thailand and the Philippines sit in the middle, offering strong recruitment potential but requiring careful EC and import management.
Sponsors that align feasibility, regulatory strategy, EC sequencing, and IMP logistics early are significantly more likely to achieve faster first-patient-in timelines and avoid costly start-up delays.
Planning clinical trials across APAC?
Clinical trial start-up in APAC is often more complex than expected. While the region offers strong recruitment potential, cost advantages, and growing research infrastructure, regulatory pathways, Ethics Committee processes, and IMP import requirements vary widely across countries. These differences can significantly impact timelines, site activation, and First Patient In (FPI).
Successful execution depends on understanding how regulatory systems and operational realities interact across markets, rather than viewing country selection in isolation. Let’s discuss your study requirements to identify the most suitable APAC countries for an efficient start-up.