Electronic Informed Consent (eICF) In Your Clinical Trial

An electronic informed consent (eConsent or eICF) is a principle in clinical research that a patient should have sufficient information before making their own free decisions about their medical care and participate in clinical research. Using electronic medium to manage informed consents makes the researchers job easy and regulatory authorities also recommend using eICF.

Did you know? As per a survey, 35% of prospective participants said that they did not enroll in a clinical trial due to a lack of understanding of the long paper informed consent forms. This indicates that there is a need for the improvement of the informed consent process.
The eICF improves accessibility of ICF content to the participants, as readers can adjust the screen, magnify, and adjust the font size, contrast, and brightness.
eConsent eliminates many roadblocks for participants and engages them by optimizing and clarifying the informed consent content.
It provides a variety of illustrations compared with the traditional process. It allows them to make more informed and take ethical decisions.
Informed consent forms in clinical trials
The informed consent (IC) process in clinical research refers to informing all the pertinent aspects of a clinical study to a potential research participant by presenting the information in a IC document.
Informed consent is an ongoing conversation between the human subject (participants) and the researchers. It begins before consent is given and continues until the end of the subject’s involvement in the research.
The informed consent process is essential to the ethical conduct of clinical trials on new medicinal products and devices. As a result, researchers have attempted to make the IC process more engaging and informative.
Presently in clinical research, there is a strong shift towards using digital technologies and this implies for eConsent too. Research shows that more than 80% of clinical trial sponsors will implement electronic informed consent (eIC) in the short term.
What is an Electronic Consent form?
Lengthy and complex paper-based informed consent documents have proven to be ineffective in understandably providing information to research participants.
An electronic Informed Consent (eIC) refers to an interactive online-based Informed Consent application that facilitates interactions over time and enables a personalized approach, adapted to research participants’ needs.
eConsent is not only an efficient and convenient process, but also provides participants the opportunity to understand and learn information using a variety of interactive methods such as text, graphics, audio, video, podcasts, interactive websites, card readers, etc. It increases participant comprehension. And consequently, better participant comprehension reduces the risk of participants drop out due to a lack of understanding of what they had consented to.
In a research, it has shown that using eConsent illustrates to
- Increase participant adherence to the study requirements and retention,
- Reduced drop-out rates and
- increased protocol adherence
Ultimately this aids the participants in benefiting from the trial treatment, which may be potentially lifesaving.
Advantages of using eConsent
Here are the following advantages of using electronic consent in clinical trials.
- reduced regulatory risk,
- improved participant satisfaction,
- increased participant comprehension
- accelerated study and site start-up
- eConsent could enable a long-term interaction with research participants by facilitating ongoing communication with the research team during and after a research study
eConsent and regulatory
As per GCP guidelines, an IC is “a process by which a subject voluntarily confirms his or her willingness to participate in a particular trial, after having been informed of all aspects of the trial that are relevant to the subject’s decision to participate.”
The US Food and Drug Administration (USFDA), In March 2015, released a draft guidance document with recommendations for clinical investigators, sponsors, and Institutional Review Boards (IRBs) on the use of electronic media and processes to obtain informed consent for clinical investigations of medicinal products.
While using this eConsent, this guidance document recommends the following.
- The e-IC must be secure with restricted access and include suitable methods to ensure confidentiality regarding the patient’s identity.
- It recommends that the subject’s information within the system must be encrypted unless it is precisely documented.
- The e-IC process should incorporate procedures to ensure that electronic documents can be archived appropriately and that all versions can be retrieved easily.
- The system should also have audit trail capability.
The three stakeholders sponsor, vendor, and IRB involve in developing e-IC
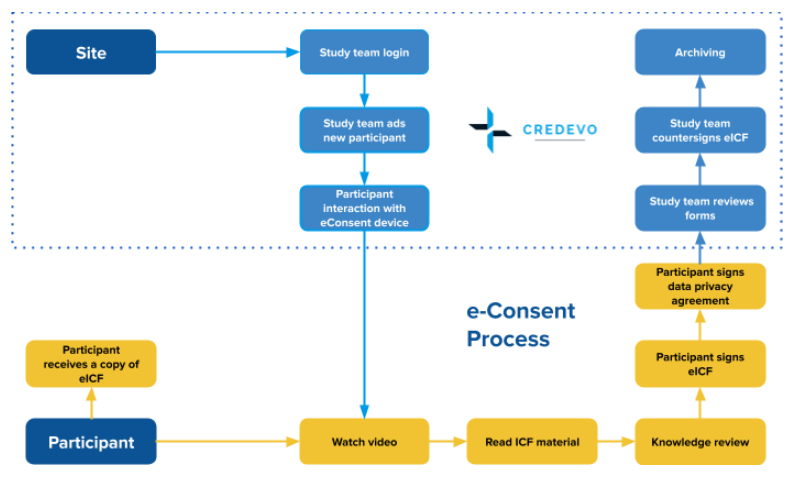
How an eConsent works?
eSignatures in eConsent
An ideal eConsent platform shall accommodate different signature types, which can be paper or electronic options compliant and in line with the country-specific regulations.
The laws for electronic signatures vary across the world. Some countries do not accept electronic signatures or require a qualified signature for informed consent purposes. If that is the case, then one can still implement eConsent with a print-to-sign signature.
This print-to-sign option would involve printing the ICF, obtaining a wet ink paper signature from the participant and a wet-ink countersignature from the study team.
E-signatures in Europe
Within Europe, electronic identification and trust services (eIDAS) EU Regulation 910 [12] addresses electronic identification and trust services for electronic transactions specific to the EU region. eIDAS highlights three electronic signature types:
- Simple electronic signature is a data in electronic form that serves a role of authentication.
- Some examples of simple signatures may include typed signatures, stylus, finger drawn.
- Advanced electronic signature is linked to the signatory in a unique and non-transferable manner as it has verification steps embedded, confirming that the person signing is indeed who he claims to be. An advanced signature is also linked to the document, preventing further modifications post the signing process.
- Qualified signature is an advanced signature created by an electronic signature device and qualified certification process.
E-signatures in USA
- The FDA also recognises electronic signatures and outlines in 21 CFR § 11 [13].
- One shall fulfil all the criteria of 21 CFR § 11. This is to consider the electronic signature equivalent to a wet ink paper signature.
- FDA recognises a variety of electronic signature methods, however it does not mandate any particular method.
Remote and audit trail in eConsent
- eConsent provides a unified platform from which one can manage the individual aspects of informed consent compliance remotely and effectively.
- eConsent shall have an electronic audit trail. This introduces new data analytics opportunities, records as all amendments to the consent documents on the system. This includes the precise date and timing, the person performing the amendment, and the reason behind the amendment.
Reconsent
When the original IC is no longer valid due to the changes in the protocol or the participant’s condition, re-consent is necessary in such cases.
Examples of such scenarios include changes in procedures, risks, potential benefits, or the worsening of the participant’s condition.
- Re-consent helps to ensure that the trial participation remains consistent with the participant’s interests.
- The sponsor sends all the amendments to the participant again, and repeats the consent process.
- If the participant chooses to consent to and sign the updated eICF, the site countersigns and the trial participation continues.
- Re-consent principles and requirements are not dependent on the consent format, paper or electronic, thus standard procedures may be followed.
Need best software management tools for your clinical trials?
Credevo provides complete guidance in clinical trial management software solutions. Provide your details below to connect with us.