Drug Development Strategy In Japan
After changes in the regulatory system, Japan has become the largest market for clinical trials in the Asia-Pacific region for both local and global studies, and Japan is now a country of choice in clinical development maps. But still, you need a proper strategy to accelerate the drug development process in Japan.
Previously it would take longer timelines for drug approvals in Japan, and one example is the Doxil drug. The Doxil drug got market approval in the year 1999 globally but, in Japan, it took ten years to obtain market approval. It would take approximately four years for drug approval in Japan but 1.5 years in the USA.
But now, things have improved drastically.
Regulatory changes
A significant regulatory change in recent years is a greater acceptance of overseas clinical data and clarity around the regulatory framework in Japan. In addition to these changes, the regulatory authority also made several efforts to reduce the study startup and review timelines.
Due to these regulatory changes in Japan, the drug approval timelines were reduced drastically to 1.5 years from 4 years. And now, for drug development, Japan has become one of the leading countries to choose from,
Along with the above regulatory reasons, here are some more reasons to choose Japan for drug development.
Top reasons to choose Japan for your clinical development
- After the US and Chinese markets, Japan is the world’s third-largest single pharmaceutical market and second-largest for prescription drugs,
- With a rapidly aging population (26.6% over 65 years old in 2016), there is an increase in the demand for healthcare medicines,
- A mature local market and renewed economy,
- Nationally-funded healthcare infrastructure with universal health insurance,
- A large and adherent patient population,
- A strong emphasis on quality and precision in clinical research,
- Incentives such as priority or conditional-approval programs for new medicines for rare diseases (high unmet needs) such as orphan diseases,
- Acceptance of foreign data and reliance on bridging studies,
- Competitive clinical trial start-up timelines,
- NDA approval timelines have been reduced significantly (as little as one year), and
- Assigning more reviewers for faster drug approvals.
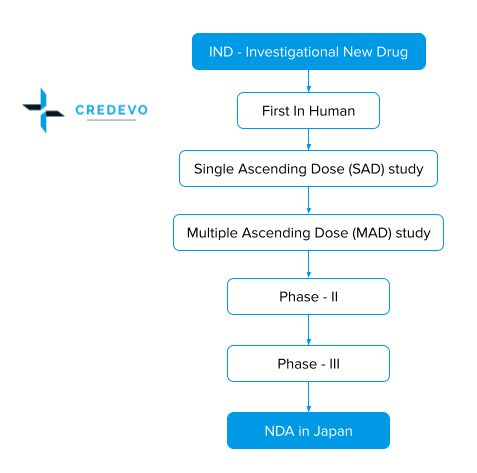
In order to conduct clinical trials and obtain drug approval, the process is typical in Japan and hence, requires some strategies. These strategies help to minimize the hurdles, delays and save both cost and time. Let’s discuss this in detail.
Need support for your drug development in Japan?
Credevo offers expertise in the drug development lifecycle and regulatory services in Japan. Connect with us to Check them out now!
Drug development strategy in Japan
In order to achieve successful drug development in Japan, one needs to follow a stepwise process below
- Understand the Japan regulatory,
- Develop a roadmap, and
- Appoint an In-country caretaker.
Understand the regulation that applies to your product
The legislation in Japan that governs pharmaceuticals and medical devices administration is the PMD Act and it regulates clinical research, manufacturing, marketing, labeling, and safety of drugs, diagnostics, regenerative medicines, and medical devices.
The sponsor needs to submit the clinical trial plan to the Ministry of Health, Labour, and Welfare (MHLW), which is the primary regulatory authority in Japan for the following
Drugs/Pharmaceuticals
- Drugs with active ingredients that differ from listed drugs in the Japanese Pharmacopoeia and drugs that are already approved for marketing.
- Substances that have the same active ingredients as those of drugs listed in the Japanese Pharmacopoeia and those drugs already approved for marketing differ in terms of routes of administration.
- Medicines that have the same active ingredients as those of drugs listed in the Japanese Pharmacopoeia and those of drugs already approved for marketing but differ from them in terms of percentage of active ingredients in the drug composition, indications and effects, and dosage and administration.
- Drug products with the same active ingredients as those of drugs that have already been approved for marketing as drugs that differ inactive ingredients from drugs listed in the Japanese Pharmacopoeia and drugs already approved for marketing, and for which the reexamination period specified under the PAL has not passed from the date of marketing approval.
- Those products are manufactured by using genetic modification technology.
- Biological products.
- Regenerative medicine products.
Medical devices/IVD devices
- Devices whose structure, usage, efficacy, effect, and performance are different from those which are already approved.
- Instruments that are medical devices approved as those that clearly differ in structure, usage, efficacy, effect, and performance from already approved medical devices and that have the same characteristics in structure, usage, efficacy, effect, and performance as those whose post-marketing usage result survey period has not passed.
Approval pathways
Understanding the Japanese regulations helps sponsors know various approval pathways for faster drug approvals and save both, cost and time. One such example is the Sakigake pathway.
Sakigake pathway
The Sakigake pathway accelerates the development of new drugs, especially innovative therapies. This pathway is similar to Breakthrough Therapy (BT) designation and fast track approach in the USA.
This pathway is applicable for breakthrough and regenerative medicines in Japan, and the advantages of this pathway include
- Substantial regulatory and scientific support from the regulators,
- Rollout NDA submissions, and
- Accelerate the review period.
In certain conditions, the Japanese regulatory may not require complete clinical trial data, rather may just ask for small studies with few patient populations to confirm the safety and efficacy. Hence, it helps to reduce the clinical development time.
Conditional early approval
Along with the Sakigake pathway, Japan has added another unique scheme known as “Conditional Early Approval,”. This scheme allows highly advanced therapies for incurable diseases without Phase 3 trial results.
Any party can utilize the “Conditional early approval” scheme in Japan. And, in this scheme, the sponsor can sell the drug in Japan with the submission of limited Japanese clinical trial data, but the drug is being developed in other countries like the USA and EU. However, the full submission should be made within 10 years or less (this period is set by the authority) with the clinical data equivalent to a Phase III trial.
So, one needs to understand the Japanese regulations to identify the right category and pathway suitable for drug development. The consultation with the PMDA before initiating any step will add more advantages.
Note: Please note that the consultation with the PMDA is a paid service.
Click here for more details about the drug approval process in Japan
Develop a roadmap as a part of the drug development strategy
To have a successful drug launch, you need to have a predefined scientific roadmap in order to avoid glitches and time delays. Once you are clear with the regulations, the next question is whether to plan your drug development in one or more countries.
One such strategy is global drug development, which means initiating regulatory and clinical trial processes in multiple regions at once when you have an idea to launch a drug in multiple regions.
Global drug development strategy
Developing drugs in more than one region will have more benefits compared with drug development within one country. This is because the clinical trial data and regulatory approval of one country give easy access to another country.
An example of global drug development: If a drug gains approval or has clinical trial data in the USA, the Japanese regulatory may consider this data and only suggest conducting bridging studies for final approval.
Top advantages of global drug development
- Prevention of unnecessary duplication of clinical trials in multiple regions,
- Plan an efficient and cost-effective drug development,
- Resolving the drug lag issue with global simultaneous drug development, and
- Data submission benefits for drug approval.
So, for a successful global drug development strategy, you need to find answers to the following questions
- What are the basic requirements to conduct a global clinical trial?
- What are the basic points to consider while designing a global clinical trial?
- How about the acceptable sample size and proportion of Japanese/Asian subjects?
- Management of concomitant medications or therapies in a global clinical trial.
- In which phase of the study can the drug development plan move into Japan?
Before you plan a global clinical trial, it is advisable to have a consultation with the PMDA as this helps to
- Get advice from the PMDA officially,
- Take advice and suggestions from the regulatory experts to design an efficient pathway, and
- Make a “binding” agreement with PMDA prior to the global clinical trial.
One such example of GCT is to submit the IND in a leading country like the USA and parallelly conduct Phase IIa studies and Phase III studies in both the USA and Japan.
Clinical studies and study designs
When you consider Japan in your development route map, then you need to keenly review the study designs. The consultation with the PMDA can play a crucial role before you finalize the study design.
Global studies, which include Japanese sites in the protocol or incorporate bridging studies from Asian countries such as Korea and/or Taiwan, are one consideration while developing a study design.
A proper study design selection helps to
- Minimizes the duplication of clinical data,
- Bridging requirements for extrapolation of FCD to a new region, and
- Enhance global development.
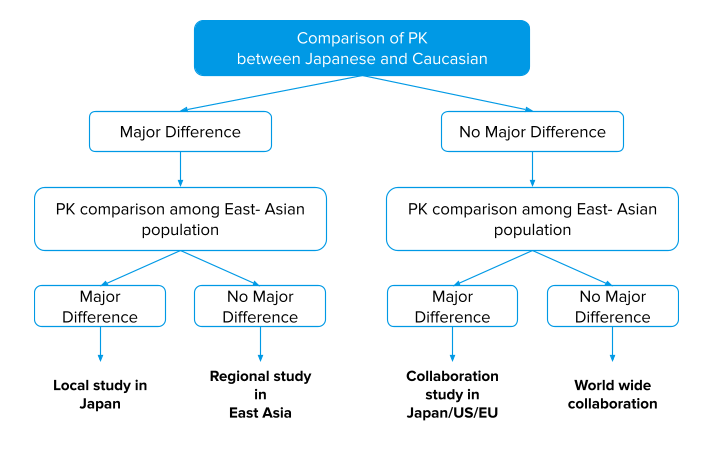
The dose-response curve differs with “ethnic groups” like, in Japan and other regions. So, most of the drugs marketed in Japan, differ from the USA, and other countries in the approved doses, and the Japanese dose is usually small.
Therefore in application dossiers, the MHLW / PMDA requires the sponsor to obtain clinical trial data in the Japanese population unless there is ground for exemption.
Tips to incorporate an efficient study design
- Needs to cover the complete clinical data package
- Adequate characterization of
- PK/PD
- Efficacy and safety
- Dose-response
- Clinical disorders evaluated using medical & diagnostic definitions acceptable in the region
- Fulfill the requirements of the bridging studies
- Ethnic considerations
- PK, PD > safety, tolerance, efficacy or dose-response
- Extrinsic (food, climate) culturally or behaviorally determined
- Intrinsic factors (genetics) greater impact on the ability to extrapolate
Management of development strategies based on the PK-profile
Adaptive design is regarded as an efficient method for clinical trials in order to increase the success rate of drug development and recently it has been actively discussed and implemented by many researchers.
Budgeting in drug development strategy
The clinical trial running costs are higher in Japan, so it is another important aspect to consider while designing a clinical trial
The higher costs are due to
- Slow patient recruitment,
- Complex payment systems, and
- Higher site costs and staffing.
Here are a few measures to reduce the costs to conduct clinical trials in Japan
Measures to reduce clinical trial costs
- Design clinical trials that require less patient recruitment (modern clinical study designs),
- Implement the best payment systems, and
- Sponsors need to negotiate with the site staff for salaries.
Patient recruitment strategies
Some strategies to improve patient recruitment
- Set up an online portal and social media and refer them to the clinical trial site.
- Collaboration with patient recruitment organizations.
- Partner with doctors, pharmacies, companies that process prescriptions, organizations that provide regular medical check-ups, and nursing care specialists.
- Utilize medical and prescription claims database.
Appoint an In-country caretaker
To conduct a clinical trial in Japan, you need to have a representative (e.g. CRO) with a strong local presence, and, in Japan, the term CRO is not very popular, rather is regarded as an In-country caretaker (ICC).
In-Country Caretaker (ICC)
An in-country caretaker is a person who handles all the clinical trial-related aspects right from registering, importing, and collecting study data.
An In-country caretaker resides in Japan (including the representative of a foreign legal person holding office in Japan) so that he carries out the necessary procedures to sponsor a clinical trial and ensure take necessary measures to prevent the occurrence or spread of hazards to public health and hygiene.
When do you need an In-country caretaker?
A sponsor can initiate, operate, monitor, and manage a clinical trial, including the preparation of standard operating procedures, complete studies on the quality; and preparation of a clinical trial protocol, etc.
However, in some conditions where the sponsor is not a local person may entrust the clinical trial operations to a CRO within the appropriate scope. And, if in the case where the sponsor is not a local person needs to appoint an in-country caretaker.
Functions of a CRO
In general, a CRO may perform any or all of the following services
- Help to plan development strategy.
- Conduct of Phase I to Phase III trials
- Preparation of approval application for.m, approval application summary, and attachment documentation as well as application.
- Post-marketing usage result survey.
- Carrying out the responsibilities of the In-country clinical caretaker and designated marketing approval holder (D-MAH).
- Trial-related services are other than those mentioned above.
- Consultation regarding development and approval/license.
- Statistical analysis.
- Quality assurance/audit and consultation regarding quality assurance/audit.
- Training services.
However, the final responsibilities concerning the quality and completeness of clinical trial data remain with the sponsor.
If the CRO acts as an in-country clinical caretaker, then it needs to take necessary measures to indemnify/compensate trial subjects in the same manner as the sponsor.
Sponsor – CRO agreement
The sponsor must enter into an agreement with the CRO, which covers the following:
- Scope of the functions and obligations.
- Procedures for the conduct of the delegated tasks.
- The provision is that the sponsor may ascertain if delegated tasks are carried out properly and smoothly in accordance with the above procedures.
- Matters concerning instructions to the CRO.
- A provision that the sponsor may ascertain if its instructions are observed and that the delegated tasks are carried out properly and smoothly in accordance with the instructions.
- Reporting related matters by the CRO to the sponsor.
- Relating to indemnification to trial subjects.
- Other necessary matters concerning the delegated tasks.
Do you need support for drug development in Japan?
Credevo offers a wide range of drug development and regulatory services in Japan. Provide the details below to receive a proposal for services.