Clinical Trial Regulatory Process – Brazil

Brazil has grown exponentially in clinical research over the last decade. During the past 3 years, clinical trials in Brazil have grown significantly, with the country becoming one of the three most dominant emerging countries, along with India and Russia, which together attract the highest number of Western companies outsourcing clinical trials.

Brazil is a large country with over 200 million inhabitants, making it the largest population in Latin America. Divided into five regions, the country is the sixth-largest pharmaceutical market in the world where regulatory compliance to clinical trials is upheld by an experienced and qualified workforce.
What are the attributes of conducting clinical trials in Brazil?
- A large, diverse, rapidly growing population,
- Hight treatment-naïve, with shortened clinical trial approval times,
- Adherence to ICH-GCP guidelines,
- High incidence of diseases prevalent in developed countries,
- Successful patient enrollment and retention rates, and
- Proximity to Western biopharmaceutical companies.
What is the regulatory for clinical trials in Brazil?
Brazilian Health Surveillance Agency (Agência Nacional de Vigilância Sanitária) (ANVISA) is the regulatory authority responsible for the review and approval of clinical trial applications for registered and unregistered drugs. ANVISA is attached to the Ministry of Health (MOH), which grants it the authority to regulate food and drug laws in Brazil.
A clinical trial application is referred to as the Drug Clinical Development Dossier or Dossier Desenvolvimento Clínico de Medicamento (DDCM) and ANVISA’s approval of the DDCM is known as a Special Notice/Bulletin or a Comunicado Especial (CE).
Do you have any queries or looking for regulatory support in various stages of drug development? write to us at [email protected] or provide your query details in the below form
How about ethics approval?
- Brazil has a centralized registration process for the ethics committee (ECs) and has a national ethics committee (CONEP) and local ethics committees (CEP).
- The Institutional ethics committee (IEC) – known as a Comitê de Ética em Pesquisas (CEP) will review and approve all clinical trial applications prior to ANVISA initiating its review and approval process.
- The National Commission for Ethics in Research (Comissão Nacional de Ética em Pesquisa) (CONEP) is the central statutory body responsible for the registration, audit, and accreditation of Institutional ethics committees (ECs), known as (Committees of Ethics in Research (Comitês de Ética em Pesquisas) (CEPs) and is the advisory body for the Ministry of Health (MOH)
- Applications with coordination or sponsorship originating outside of Brazil require an additional EC review by the National Commission for Ethics in Research (Comissão Nacional de Ética em Pesquisa) (CONEP)
Is there any clinical trial registry?
- Plataforma Brasil is a national and unified registry for research involving human participants.
- The platform also represents the review and approval processes of both the CEP and CONEP.
Research applications can be tracked from submission to final approval by the CEP, and when necessary, by CONEP. - it is mandatory for all applicants to register their clinical trials with the World Health Organization’s (WHO) ICTRP or other registry recognized by the ICMJE.
What are the requirements for ANVISA approval?
- Clinical research protocol
- Proof of Deposit Health Surveillance Rate (TFVS) (tax payment imposed on individuals and companies engaged in clinical research)
- Drug development plan
- Certified copy of the clinical agreement (contract or statement) that has been written, dated, and signed by the sponsor or his/her CRO
- Ethics in Research Committee (ERC) (also known as a CEP) opinion issued for the first clinical trial center
- Investigator’s Brochure (IB)
- Summary of investigational product’s (IP’s) safety aspects based on previous research in humans
Information on any discontinued development or withdrawal of IP - IP dossier
- Specific dossier for each clinical trial to be conducted in Brazil
- Proof of clinical trial registration with the World Health Organization’s (WHO) International Clinical Trials
- Registry Platform (ICTRP) or other registry recognized by the International Committee of Medical Journal Editors (ICMJE)
How to get approval?
- The Clinical Trial Application and associated documents (including the protocol, investigator brochure, informed consent form, and sponsor and institutional declarations), as well as all documentation provided to the CONEP/CEP System, must be translated into Portuguese
- The Principal Investigator (PI) is responsible for submitting an application via the online Plataforma Brasil to the respective Institutional EC (CEP), and, if applicable, to the National Commission for Ethics in Research (Comissão Nacional de Ética em Pesquisa) (CONEP)
- For the multicenter clinical trial, the principal investigator (PI) shall submit a list of the participating institutions and the associated protocols as part of the research protocol package sent to the CEP for review.
- The CEP will review the protocol documentation for completeness which should be accomplished within 10 days following submission and shall issue an initial report 30 days from the date the protocol documents are fully accepted for review.
- CONEP (for which sponsorship or coordination originate outside of Brazil) must also review applications and shall issue its initial report for this additional review within 60 days from the date the documentation was accepted.
- Clinical Research Coordination on Drugs and Biologicals (Coordenação de Pesquisa Clínica em Medicamentos e Produtos Biológicos (COPEC) at ANVISA’s office is responsible for conducting the review of clinical trial applications.
- All communication between the research center, the principal investigator, and the ethical committee system should be done through the online platform
- Once approved by the Ethics committee application can be forwarded to ANVISA
ANVISA’s approval of a clinical trial application is dependent upon obtaining proof of the EC’s (CEP’s) approval - Application includes
- A hard copy of the clinical trial application (1) and
- Electronic copy on CD-ROM in Adobe Acrobat PDF, Microsoft Word, or Open Document file format. (1) (The electronic documents should also have text searching capability).
- The evaluation process by ANVISA is usually divided into two phases:
- Ensure that all the required documentation for the clinical trial application is included;
- Technical review of the application.
- Upon receipt of the DDCM, ANVISA has 90 calendar days to evaluate the application. If ANVISA fails to issue a response in 90 days after receipt, clinical development can begin as long as all of the ethical approvals have been obtained.
- The review and approval process by ANVISA typically takes an average of four to eight weeks.
- Once approved from ANVISA the sponsor or CRO is required to obtain an import license from ANVISA for the shipment of the investigational product (IP) to be used in the trial.
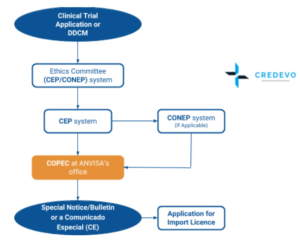
- DDCM: Drug Clinical Development Dossier or Desenvolvimento Clínico de Medicamento,
- CEP: Institutional ethics committees (Comitês de Ética em Pesquisas)
- CONEP: National Commission for Ethics in Research (Comissão Nacional de Ética em Pesquisa)
- ANVISA: Brazilian Health Surveillance Agency (Agência Nacional de Vigilância Sanitária)
What is the fee structure?
- Applicants shall pay a Proof of Deposit Health Surveillance Rate (TFVS) fee to submit a clinical trial application to the Brazilian Health Surveillance Agency (Agência Nacional de Vigilância Sanitária) (ANVISA).
- The amount of the TFVS fee depends on the size of the organization submitting the application.
- The fee ranges from R$1,421.70 to R$28,433.93 BRL.
- There is no fee charged for EC approval
What is the approval timeline?
- ANVISA has 90 calendar days to evaluate a submitted DDCM; no response from ANVISA means the trial has been approved and can commence if ethical approval has also been received.
- For clinical development of biological products including vaccines, and Phases 1 and 2, the evaluation time period is 180 days. A response must be received before the study can begin.
Generics
- In order to prove the safety and efficacy of generics – Branded and Non-Branded drugs, Clinical Trials are not necessary to perform, provided the clinical trials have already been performed on reference drugs and prove that the generic drugs are bioequivalent to reference drug based on the previous data.
- If the applicant can prove bioequivalency, ANVISA will assume that the branded and/or non-branded generic drug is safe and effective by relying on the clinical data that were evaluated during the registration of the new drug.
- The applicant seeking approval for Generic drug must submit documents of bioequivalence/Bioavailability study reports, details of drugs, etc
- All the Bioequivalence studies (BE) conducted by a certified contract research organization should be submitted to ANVISA for registration of generic and biosimilar products and CRO must comply with GCP (Good Clinical Practice) and GLP (Good Laboratory Practice)
- For BE studies, the sponsor should get approval from CONEP before initiation.
Clinical trial statistics
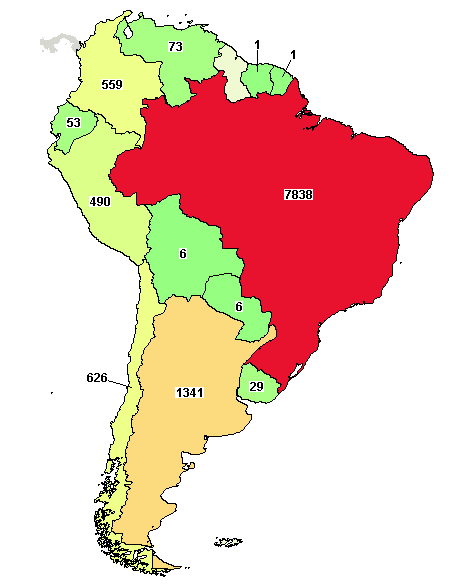
A total of 7,838 clinical studies were registered to date with 1,469 Ongoing studies.
The major therapeutic areas are
- Heart and Vascular Disorders
- Oncology
- Immune Disorders
- Infection and communicable disease
Need Support in Brazil or Have questions?
We’d love to help you conduct clinical trials in Brazil.
Connect with experienced and resourceful sites, services providers, and experts in Brazil.
Provide your details below.
References
- https://www.languageconnections.com/addressing-issues-affecting-clinical-trials-in-brazil/
- http://www.jforcs.com/wp-content/uploads/2015/10/Market-2-Brazil.pdf
- https://clinregs.niaid.nih.gov/country/brazil#authorizing_body
- https://www.nihcollaboratory.org/sites/CbyC/Pages/CSIGovAg.aspx?Country=Brazil
- https://clinicaltrials.gov/ct2/results?recrs=ad&cntry=BR&map=SA