Want to Conduct Clinical Trials in the United States? Here’s what you need to know – Part 2

The United States is the region that conducts most clinical trials in the world and is the flag bearer for most clinical research studies requires considering many factors and areas.

Starting with some salient points on Clinical trials in the United States.
In the first part of this series of articles on clinical trials in the United States, we have seen the regulatory situation, clinical sites, and investigators. In this article, we will understand the regulatory aspects of medical devices and other aspects of clinical trials in the USA.
Salient features of Clinical trials in the United States
- Language for a clinical trial application: English
- Process of regulatory & EC approval: Parallel
- Clinical trial registration: Required
- Studies on minors: The state determines age.
- Export of specimens: Allowed
Regulatory for medical devices Clinical trials in the United States
What are the regulatory bodies for medical devices in the USA?
FDA’s Center for Devices and Radiological Health (CDRH) is responsible for regulating firms that manufacture, repackage, relabel, and/or import medical devices sold in the United States.
CDRH regulates radiation-emitting electronic products (medical and non-medical) such as lasers, x-ray systems, ultrasound equipment, microwave ovens, and color televisions.
- Medical devices are classified into Class I, II, & III, and the regulatory control increases from Class I to Class III.
- The device classification regulation defines the regulatory requirements for a general device type.
- Class-I devices are exempt from premarket notification.
- Class-II devices require premarket notification.
- Class-III devices require premarket approval.
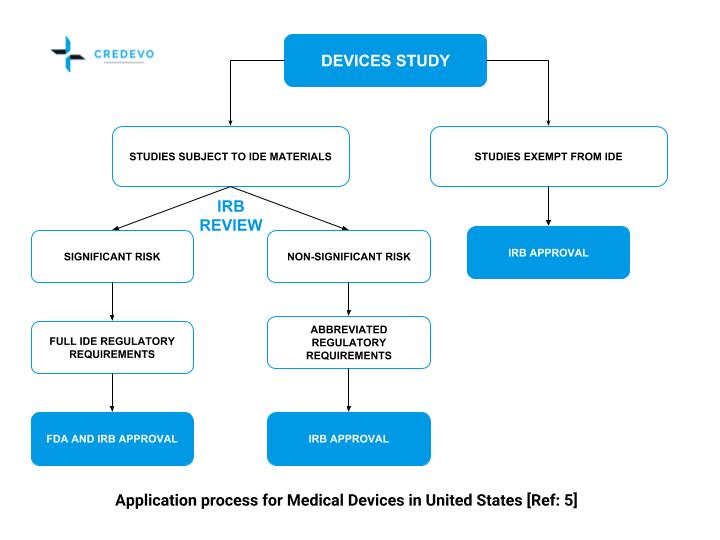
Which regulations apply to the clinical investigation of a medical device?
The regulations in clinical studies of medical devices must comply with
- FDA’s human subject protection requirements,
- Institutional Review Board (IRB) requirements,
- Investigational Device Exemptions (IDE) requirements,
- Financial Disclosure for Clinical Investigator’s requirements, as well as
- Any other applicable regulations, including pertinent regulations.
Need support for your clinical trials in the United States?
Credevo offers expertise in clinical trial regulations, drug product registration, and many more services in the United States. Provide the details of your requirement in the form below to connect with us.
Regulatory changes in IRB Requirements for medical device clinical trials in the United States
The organization that plans to conduct the particular type of clinical trials may rely on central rather than for reviews required by the US Food and Drug Administration.
- The FDA’s Center for Devices and Radiological Health (CDRH) has issued a technical amendment allowing sponsors to conduct clinical studies for Humanitarian Devices.
- Exemption (HDE) devices utilize central IRBs to review their activities; previously, it was necessary for the clinical trial sponsors and sites to undergo review by local IRBs.
- The allowance for central IRB review applies to Investigational Device Exemption (IDE) studies and multi-site clinical studies.
- This change is to bring drug and device regulations more closely in line with one another, according to the FDA.
When the regulations don’t apply?
- An FDA-approved device to test a physiologic principle where no data is collected about the device,
- An FDA-approved device to address a research question and no data is collected about the device, or
- Using an FDA-approved device for clinical purposes (e.g., monitor a side effect, measure treatment progress),
- As long as there is no intent to collect safety or effectiveness data or develop the device for marketing. (use of an MRI to measure a clinical outcome in a study that has nothing to do with the MRI itself).
The approval process for medical device clinical trials in the United States
Various types of drug applications
Investigational New Drug (IND)
As per Federal Law, a drug requires marketing approval before transportation and distribution across state lines.
- New Drug Application (NDA),
- Abbreviated New Drug Application (ANDA),
- Biologic License Application (BLA).
What is the submission process?
- In the United States, the sponsor should submit an investigational new drug application (IND) for FDA review and authorization.
- Whether a drug requires an IND or not before marketing primarily depends on the intent of the investigation and the degree of risk associated with the use of the drug.
- Institutional ethics committee (EC) review of the clinical investigation may be conducted in parallel with the FDA review of the IND. However, EC approval must be obtained before the sponsor is permitted to initiate the clinical trial.
Clinical trial registry
- Either the sponsor or the principal investigator (PI) designated by the sponsor should register electronically with the Clinicaltrials.gov databank.
- The sponsor/PI must register 21 calendar days after the first human participant is enrolled in a trial.
Electronic regulatory submission and review
The Electronic Common Technical Document (eCTD) is the standard, accepted electronic format for the following submission types:
- Investigational New Drug Application (IND)
- New Drug Application (NDA)
- Abbreviated New Drug Application (ANDA)
- Biologics License Application (BLA)
- Master files: Drug Master File (DMF) and Biologics Master File (BMF)
- Emergency Use Authorization (EUA)
If the applicant submits the data electronically, it is easier for FDA to review data, approve new drugs, and monitor drugs after they go on the market.
When a clinical trial can get FDA assistance?
Drug developers can seek help from FDA at any point during the drug development process, including
- Pre-IND application, to review FDA guidance documents and get answers to questions that may help enhance their research.
- After Phase 2, to obtain guidance on the design of broad Phase 3 studies.
- Any time during the process for assessment of the IND application.
Regulations for the import and export of medicinal products for clinical trials in the United States
The Centre for Drug Evaluation and Research (CDER) regulates the import and export of medicinal products.
- Foreign drug manufacturers who wish to import drugs into the United States must register with the FDA.
- The Federal Food, Drug, and Cosmetic Act prohibits the import or export of unapproved new drugs, meaning any drug that has not been manufactured with FDA approval.
- Imported drug products are subject to inspection by the US Bureau of Customs and Border Protection, so the FDA may refuse admission of any drug that appears to be unapproved, misbranded, or adulterated.
When to initiate a trial?
Applicant can initiate the clinical trials immediately after the 30-day approval, after receiving IND and Ethics committee approval. The sponsor or the Principal investigator designated by the sponsor should register the Clinical Trials with clinicaltrial.gov.
What are the reporting and disclosure requirements?
Food and Drug Administration requires all applicable clinical trials to register and report results on a government-administered website (www.clinicaltrials.gov). If a clinical trial involves Phase I studies of a drug, then the results submission is not required.
FDA obligations with respect to clinical trial subjects
The FDA requires that clinical trial subjects be given adequate information and time to make an informed decision on whether to participate in a clinical trial and ask questions and have those questions answered.
Insurance requirements for clinical trials
- In the United States, human clinical trial liability insurance is not mandatory. But the multi-country trials are likely to require multiple insurance policies.
- Hence it is recommended that companies consider brokers and lawyers familiar with jurisdictional requirements to enter into sponsorship agreements.
What are the data protection concerns?
Data protection issues considered while conducting clinical trials
Most clinical trials today are designed according to International Conference on Harmonisation (ICH) guidelines with multiple jurisdictions in mind.
We can consider data protection into two basic categories
- Data integrity and retention, and
- Patient privacy.
With regard to data integrity and retention, the FDA periodically issues guidance for industry documents on its website, which should be constantly monitored for updates.
and finally
Why USA is preferred destination for clinical trials, in numbers?
The country contributes the most clinical trial participants in the world. The USA (0.35) makes up a little more than 4% of the world’s population.
- According to ClinicalTrials.gov, as of April 2023, there are over 378,000 clinical trials registered worldwide, with the USA accounting for over 170,000 trials, or approximately 45% of all registered trials.
- This is significantly more than any other country, with the next highest being China with over 19,000 registered trials.
- The USA’s dominance in clinical trials is due in part to its large and diverse patient population, which makes it easier to recruit participants for clinical trials.
- According to the US Census Bureau, the USA has a population of over 331 million people, making it the third most populous country in the world.
- Additionally, the USA has a diverse population, which is important for clinical trials that require participants from different racial and ethnic backgrounds.
Click here for more information to Conduct Clinical Trials in the U.S.? Here’s what you need to know – Part 1
Need Support in the USA or Have questions?
We’d love to help you conduct clinical trials in the United States.
Connect with experienced and resourceful sites, services providers, and experts in the United States.
Provide your details below.