Want to Conduct Clinical Trials in the United States? Here’s what you need to know – Part 1

The United States has consistently emerged as a leading site for clinical research. Half of the total clinical trials conducted worldwide are being conducted in the United States. Most of the research pharma companies prefer to perform their clinical trials in the United States due to the reasons such as well-established medical infrastructure, fast approval timelines, agile regulatory framework, widely accepted clinical trial data, and many more.

Clinicaltrials.gov registry, which serves as an effective reference to see clinical trials conducted worldwide, provides interesting statistics regarding U.S. clinical trials.
Statistics of clinical trials in the United States
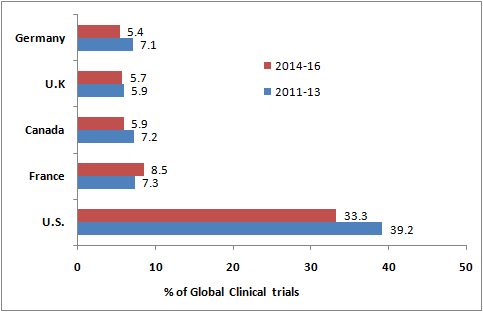
- A glimpse into the studies registered between 2011 and 2013 and further between 2014 and 2016 shows that the U.S. and Western Europe are the major hubs to conduct clinical trials.
- The United States had the highest number (33.3%) of clinical trials registered in 2014-2016 and was followed by France (8.5%), Canada (5.9%), United Kingdom (5.7%), and Germany (5.4%).
- Together, these countries are located in the traditional regions of North America and Western Europe, hosting 58.8% of all clinical trials.
Top reasons to choose the United States and Western Europe for clinical trials
The compelling reasons to choose the United States and Western Europe for clinical trials are
- Availability of thousands of qualified, inspected and verified investigators
- The robust and easy regulatory system in place
- Great professional clinical research culture
Leading global pharmaceutical companies conduct the majority of their clinical trials in the United States and Western Europe.
When you plan to conduct the clinical trial in the United States, then here are several things to consider.
- Regulatory for conducting clinical trials in the United States, and
- Sites and Investigators for conducting clinical trials in the United States.
The first thing to consider is the regulatory requirements and necessary approvals.
Need support for your clinical trials in the United States?
Credevo offers expertise in clinical trial regulations, drug product registration, and many more services in the United States. Provide the details of your requirement in the form below to connect with us.
Regulatory for conducting clinical trials in the United States
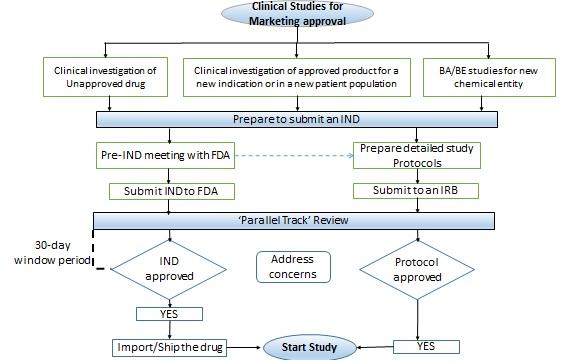
Conducting clinical studies in the U.S
- An investigational new drug (IND) application is usually submitted to propose a clinical investigation of an unapproved drug, or an approved product for a new indication or in a new patient population.
- If the test product contains a new chemical entity or involves a radioactively labeled drug product or a cytotoxic drug product, then an IND is also required to conduct a bioavailability or bioequivalence (BA/BE) study.
- The U.S. FDA responds to an IND application within 30 days.
- if the FDA has any concerns, in that case, the sponsor needs to respond to the queries and can start the study only after a written notification from the FDA to do so.
- In case, if there is no response from the FDA in the 30 day window period, the sponsor can start the clinical development program, as described in the IND on the 31st day provided, the institutional review board (IRB) approval is in place.
The figure depicts an overview of the conduct of clinical studies for marketing approval in the United States
Supportive regulatory framework for generic products
- The Hatch-Waxman Act, 1984, provides a supportive framework to the generics industry and the resultant competition to their brand counterparts.
- The generics market has grown to account for nine out of 10 prescriptions dispensed in the U.S. today by abbreviating processes and reducing sponsor cost.
- For generics, the 505(j) is the approval route.
- Using unapproved versions of approved drug products, BA/BE studies can be conducted without submission of an IND [21 CFR 320.31(b) and (d)].
- Further, 505(b)(2) NDA is like a hybrid between the full application 505(b)(1) NDA and the 505(j) ANDA. (NDA- New drug application; ANDA- Abbreviated new drug application).
- The provisions of 505(b)(2) allows some information required for NDA approval, to come from studies not conducted by or for the applicant. This has greatly reduced the cost to the sponsor and allows for a faster route for drug approval.
Speeding up the clinical development program
- The FDA encourages all potential drug sponsors to utilize the Pre-IND Consultation Program.
- This can be used to discuss the drug development path and suitable regulatory pathway for marketing authorization upfront with the FDA.
- A sponsor may simultaneously apply for the protocol(s) (as mentioned in the IND) approval by the IRB.
- As required by 21 CFR 56.106, IRB in the United States that reviews FDA-regulated studies should be registered with the Office for Human Research Protections (OHRP).
- The Department of Health and Human Services (HHS) maintains the IRB registration information.
- There are more than 10,000 IRBs that are registered with the OHRP IRB registration system.
- A sponsor may seek IRB approval before an IND application however, recruitment and enrollment do not begin before both effective IND and IRB approval are secured.
Clinical trial drug product shipment
- The sponsor may want to transport or distribute the clinical study drug across the state borders. However, according to the current U.S. federal laws, only drugs with marketing approval can be transported or distributed across state lines.
- An IND application provides an exemption to this law.
- It is only required that such a product intended for human use should be labeled with the statement “Caution: New Drug–Limited by Federal (or the United States) law to investigational use.”
If you’re familiar with the regulatory requirements and are confident to comply with them, the next thing to consider is the availability of sites and investigators in the United States.
Sites and Investigators for conducting clinical trials in the United States
Investigators in the United States are some of the most informed, experienced, qualified, and willing to perform investigators in the world. It also helps that they work in one of the most advanced countries.
Available therapeutic areas
The United States is home to patients from a number of therapeutic areas ranging from some of the common ones like oncology to even rare indications like Acute unilateral vestibulopathy.
As per an article published in 2013 (https://www.ncbi.nlm.nih.gov/pubmed/23842577) on disease burden in the U.S., diseases with the largest number of YLLs (Years of life lost due to premature mortality) in 2010 were ischemic heart disease, lung cancer, stroke, and chronic obstructive pulmonary disease. While, diseases with the largest number of YLDs (Years lived with disability) in 2010 were low back pain, major depressive disorder, other musculoskeletal disorders, neck pain, and anxiety disorders; neuropsychiatric disorders are now considered as the leading cause of disability in the U.S., followed by cardiovascular and circulatory diseases and neoplasms (https://www.nimh.nih.gov/health/statistics/disability/us-leading-categories-of-diseases-disorders.shtml).
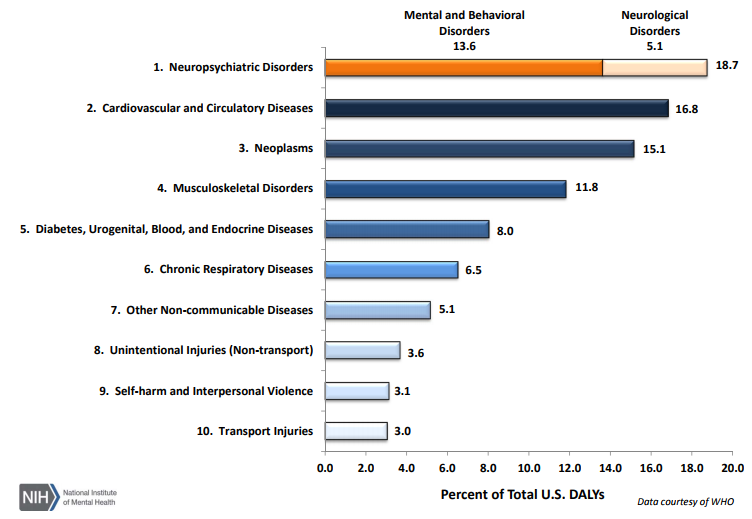
The U.S. can be a great place to go for one is looking for investigators for clinical trials in oncology, cardiovascular diseases, metabolic diseases like diabetes, psychopathic disorders, neurology, and other leading disorders listed above,
Numbers of sites and investigators
As per a book from NIH (https://www.ncbi.nlm.nih.gov/books/NBK50886/) published in 2010, a total number of investigators in the United States was 14000, although this seems to be the figure for clinical trials active at that time. The number has shown to be more based on the trials posted on clinicaltrials.gov.
Credevo has access to about 25000 investigators in the U.S. These investigators work in about 40 therapeutic segments and come from sites managed by their own team or by other site management organizations. Some of the sites are quite well managed and work in a group of few to more than 1000 per site.
Need Support in the USA or Have questions?
We’d love to help you conduct clinical trials in the United States.
Connect with experienced and resourceful sites, services providers, and experts in the United States. Provide your details below.
2 thoughts on “Want to Conduct Clinical Trials in the United States? Here’s what you need to know – Part 1”
Comments are closed.