Rivaroxaban Bioequivalence Study Protocol
Rivaroxaban (Xarelto) generates around $6 billion annually with indications spanning stroke prevention, VTE treatment/prevention, and cardiovascular risk reduction within the $20+ billion global anticoagulant therapeutics market. The rising prevalence of atrial fibrillation and venous thromboembolism drives market expansion and chronic therapy needs. With one of the most BE-friendly anticoagulant profiles, featuring predictable pharmacokinetics and low variability enabling rapid, low-risk development, early movers dominate the market, with the first couple of generic entrants capturing 70-80% of the initial generic share. This ready-to-use protocol provides an instant 6-8 week head start in one of the decade’s largest anticoagulant generics opportunities.
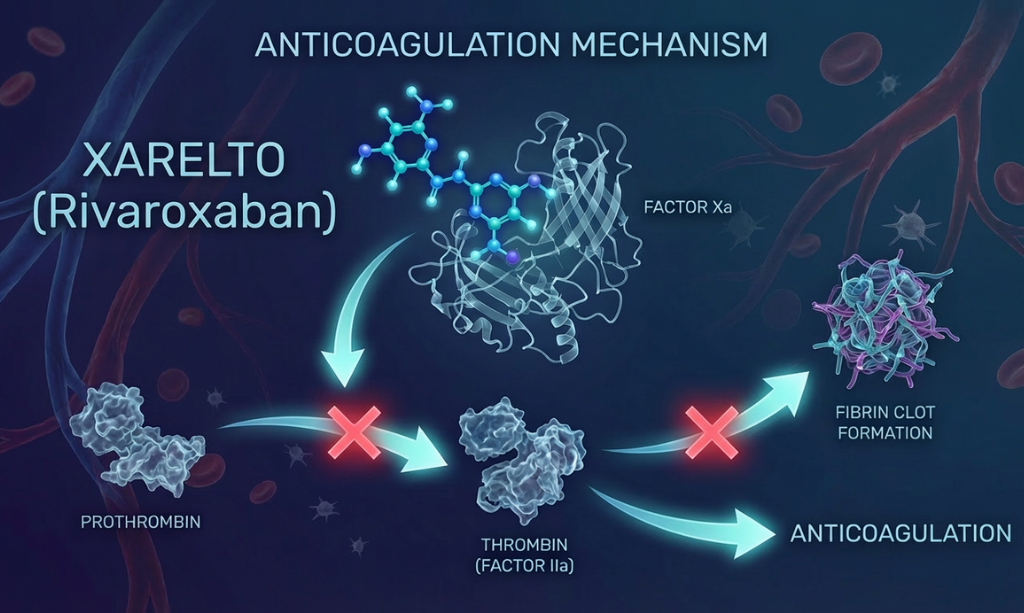
Overview: Rivaroxaban (Xarelto) is used for stroke prevention in atrial fibrillation, deep vein thrombosis (DVT) and pulmonary embolism (PE) treatment and prevention, and cardiovascular risk reduction in patients with coronary or peripheral artery disease, providing convenient once or twice daily dosing with predictable anticoagulation and no routine monitoring required. It is a direct, highly selective oral Factor Xa inhibitor that blocks the coagulation cascade to prevent thrombin generation and clot formation. A bioequivalence study confirms that generic formulations are equivalent to the reference product in terms of systemic exposure and pharmacokinetic profile.
Download the complete, submission-ready BE protocol with all technical details
What This Protocol Includes
- Study Synopsis
- Study Design Narrative
- PK Parameter Definitions (AUC, Cmax, Tmax, t½)
- Safety and AE/ SAE Reporting
- Sampling Time Points
- Schedule of Assessments
- Washout period
- Scientific Rationale & Regulatory Justification
- Dosing & Administration
- Inclusion/ Exclusion Criteria
- Sample Handling
- Statistical Considerations

Read More
Explore our comprehensive collection of ready-to-use bioequivalence study protocols designed to accelerate your generic drug development and streamline regulatory approval pathways. Each protocol is meticulously crafted to meet FDA and EMA requirements, providing detailed study designs, pharmacokinetic parameters, and statistical methodologies tailored to each molecule’s unique characteristics.
Study Design Framework
Each protocol includes a molecule-specific study design, developed with careful consideration of pharmacokinetics, variability, dosage form, and applicable regulatory guidance. This includes:
- Study type and design
- Dose strength and administration conditions
- Washout period justification
- Risk–benefit assessment
The study designs are structured to reliably capture intra- and inter-subject variability and to support robust bioequivalence conclusions.
Comprehensive Pharmacokinetic Framework
Each protocol provides a detailed pharmacokinetic (PK) assessment plan, including:
- Defined primary and secondary PK parameters
- Blood sampling schedules optimized for both absorption and elimination phases
This framework ensures alignment with regulatory expectations for accurate and reliable exposure assessment.
Statistical Considerations Aligned with Regulatory Standards
Protocols include a clearly defined statistical analysis plan, covering:
- Bioequivalence acceptance criteria
- Appropriate statistical models
- Handling of dropouts
- Population definitions
The statistical approach is designed to meet FDA and EMA review standards.
Protocol Overview & Regulatory Positioning
Each protocol is a fully developed BE study protocol intended to support ANDA and global generic submissions, and includes:
- Alignment with FDA Product-Specific Guidance
- Design to demonstrate bioequivalence between the Test product and Reference Listed Drug (RLD)
- Compliance with:
- ICH E6 (R3) Good Clinical Practice
- FDA regulations
- EMA bioequivalence guidelines
- Structured for IRB/ IEC submission with minimal customization
- Suitable for U.S. and international regulatory filings
Safety Monitoring & Risk Management
Each protocol incorporates a comprehensive safety assessment plan, including:
- Monitoring and reporting of adverse events (AEs) and serious adverse events (SAEs)
- Vital signs, ECGs, and clinical laboratory evaluations
- Molecule-specific safety considerations
- Subject-level and study-level stopping rules
- End-of-study and follow-up safety assessments
Operational Readiness
Our protocols go beyond regulatory requirements and include practical execution details, such as:
- Screening and eligibility assessments
- Check-in, dosing, and confinement procedures
- Activity, dietary, and compliance restrictions
- Sample handling and bioanalytical method compliance requirements
This ensures smooth implementation at clinical sites or CROs.
Quality Assurance & Inspection Readiness
Each protocol incorporates quality and compliance elements to support inspection readiness, including:
- Monitoring and audit preparedness
- Source documentation expectations
- Data handling, archival, and retention requirements
- Traceability aligned with FDA and EMA inspection standards
Home > Clinical Study Design > Ready-to-Use Bioequivalence Protocols for High-Value Generics > Bioequivalence Study Protocols > Rivaroxaban