Medical Device Market Approval Process in the United States

The United States remains the largest medical device market in the world with $156 billion which is 40 percent of the global medical device market in 2017 and By 2023, it is expected to grow to $208 billion

The medical technology industry (commonly referred to as medical devices) consists of articles, instruments, apparatuses, or machines that are used in the prevention, or for detecting, measuring, restoring, correcting, or diagnosis or treatment of illness or disease or modifying the structure or function of the body for some health purpose.
The regulatory body for medical devices in the United States
The U.S. The Food and Drug Administration’s Center for Devices and Radiological Health (CDRH) oversees medical devices marketed in the United States.
How are medical devices classified?
Medical devices are classified into three categories based on the associated risk, namely: Class I, II, and III.
- Class I devices will have the least associated risk while
- Class III devices will have the highest associated risk. Accordingly, regulatory control surges from Class I devices to Class III devices.
Need support for your medical device registration in the United States?
Credevo offers expertise in clinical trial regulations, drug product & medical device registrations, and many more services in the United States. Provide the details of your requirement in the form below to connect with us.
US FDA Regulations for various classes of drugs
- Most Class I devices are exempted from 510(k) premarket notification submission,
- While most Class II devices are submitted for premarket notification.
- On the other hand, Class III devices need to go through the Premarket Approval Application (PMA) and other class III devices, which are exempted from PMA must submit a 510(k) notification to FDA.
Depending on several factors, including the potential risk that a device poses to patients, the CDRH has varying levels of scrutiny it applies to new products being introduced.
- All the medical device manufacturers and distributors must register their organization with the FDA to sell their devices.
- All the establishments should be registered electronically using the FDA Unified Registration and Listing System (FURLS system).
How foreign manufacturers can market their devices in the US?
Foreign manufacturers must have a U.S. Agent.
- They may hire a U.S. agent as their official correspondent.
- They only need to provide primary details of the U.S. agent such as name, number, email ID, and physical address.
Types of regulations
Premarket Notification 510(k)
- Premarket notification is required for most, but not all medical devices.
- If a device requires the submission of premarket notification, it cannot be commercially distributed in the United States until it receives authorization from the FDA.
Premarket Approval (PMA)
- For medical devices that pose a high threat to patients health should undergo Premarket Approval (PMA) risk-based evaluation process
- Manufacturers of Class III devices (and devices that are not substantially equivalent to Class I or Class II) are required to submit a premarket approval application.
- The manufacturer cannot start any marketing activities before receiving premarket approval.
- FDA shall not take more than 180 days to issue a decision on whether a submitted PMA has been approved or rejected, however, this may take even longer times.
Investigational Device Exemption (IDE)
Investigational Device Exemption (IDE) allows manufacturers to use the device in question in clinical studies to collect evidence that proves its general safety and effectiveness.
- All studies performed on devices associated with a significant risk shall be approved by the FDA and the Institutional Review Board (IRB) before manufacturers initiate clinical trials and start collecting relevant data.
- Clinical studies on devices of lesser risk must be approved only by the IRB.
Quality system regulation
The Quality System Regulation includes requirements related to methods, controls, and facilities used for the designing, manufacturing, labeling, packaging, storing, purchasing, installing, and servicing of medical devices.
The quality system enforced by the FDA is also referred to as Current Good Manufacturing Practice (CGMP)
Labeling Requirements
It is mandatory to apply labels on each device. The labeling contains labels and literature in the form of descriptions and information that accompanies the device usage.
This regulation specifies the requirements in the below form
- General Device Labeling
- Use of Symbols
- In Vitro Diagnostic Products
- Investigational Device Exemptions
- Unique Device Identification
- Good Manufacturing Practices
- General Electronic Products
Medical Device Reporting (MDR)
In several cases where a medical device may cause a death or a serious injury or in case of a certain device malfunction, it is required to report the FDA for the same.
Here are the reporting rules to be followed by manufacturers, importers, and medical facility owners:
- Manufacturers: Need to report to the FDA using 3500A form when they identify that their device caused death or serious injury. They are also bound to inform the FDA when some serious malfunction happens which may cause a death or a serious injury and there is a possibility that this may happen again.
- Importers: Importers need to inform the FDA as well as manufacturers in case of death or injury. While for device malfunction, they need to report to manufacturers only.
- Healthcare Facility: When a hospital, ambulatory surgical facility, or nursing home, etc where a medical device is being used detects that some device would have caused a death, they need to inform FDA and manufacturer as well.
How to identify to which class your drug belongs?
Determine the classification of your medical device or In Vitro Diagnostic (IVD) device by searching the FDA classification database using relevant search terms, or by identifying another (predicate) device with the same intended use and technology.
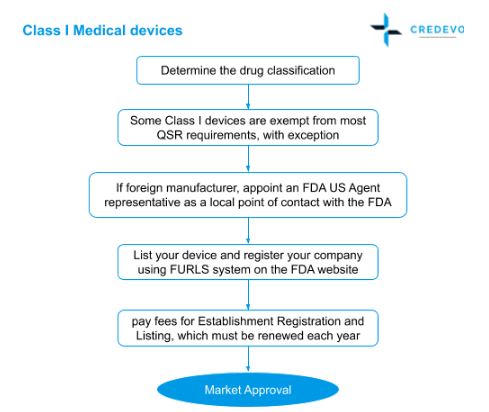
FDA approval process for Class 1 devices
- Some Class I devices are exempt from most QSR requirements, with exceptions
- If the manufacturer is a foreigner and has no local presence, he shall appoint an FDA US Agent representative as a local point of contact with the FDA.
- List the device and register your company using the FURLS system on the FDA website;
- pay fees for Establishment Registration and Listing, which must be renewed each year.
- Now the manufacturer can sell the devices in the US.
- The company and device registration status will be listed on the FDA website.
- The device’s marketing authorization does not expire as long as no changes are made to the device design, intended use, etc.
Process flow
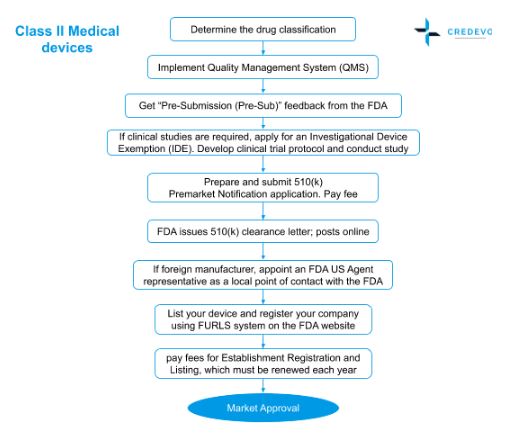
FDA approval process for Class II devices
- Determine the classification of your medical device or In Vitro Diagnostic (IVD) device by searching the FDA classification database using the relevant search term
- Implement a Quality Management System (QMS) that meets FDA Quality System Regulation (QSR)
- Innovative Class II and all Class III devices will likely require clinical studies.
- Sponsor shall get get “Pre-Submission (Pre-Sub)” feedback from the FDA
- If clinical studies are required, the sponsor shall apply for an Investigational Device Exemption (IDE). Develop clinical trial protocol and conduct study
- Prepare and submit a 510(k) Premarket Notification application. Pay required fee
- FDA issues 510(k) clearance letter; posts online
- At this time, the sponsor must be in full compliance with QSRs
- The FDA will not inspect Class I or II device manufacturers for compliance prior to device registration, but once registered, the FDA may conduct random inspections and can issue a Form 483 for non-compliance
- If the manufacturer is foreigner, shall appoint an FDA US Agent representative as a local point of contact with the FDA
- List the device and register your company using FURLS system on the FDA website; pay fees for Establishment Registration and Listing, which must be renewed each year
- Now the manufacturer can sell the device in the US.
Process flow
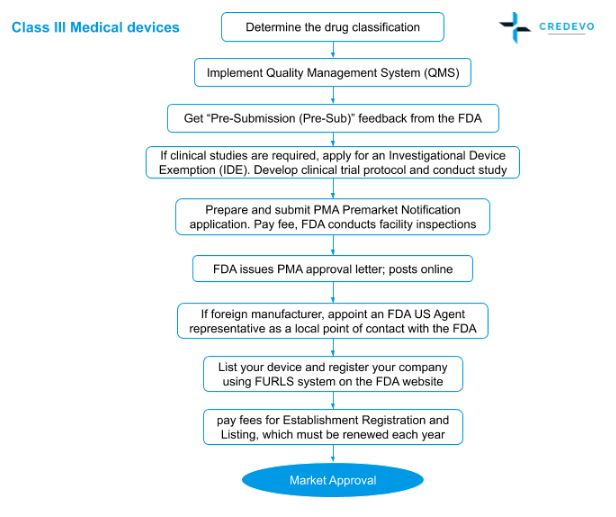
FDA approval process for Class III devices
- Determine the classification of your medical device or In Vitro Diagnostic (IVD) device by searching the FDA classification database using the relevant search term
- Implement a Quality Management System (QMS) that meets FDA Quality System Regulation (QSR)
- Innovative Class II and all Class III devices will likely require clinical studies.
- Sponsor shall get get “Pre-Submission (Pre-Sub)” feedback from the FDA
- If clinical studies are required, the sponsor shall apply for an Investigational Device Exemption (IDE). Develop clinical trial protocol and conduct study
- Prepare and submit a PMA Premarket Notification application. Pay required fee
- FDA conducts facility inspections of the manufacturer and all major suppliers involved in the design and production of your device. All parties must be compliant with FDA QSR
- FDA issues PMA approval letter; posts online
- At this time, the sponsor must be in full compliance with QSRs
- The FDA will not inspect Class I or II device manufacturers for compliance prior to device registration, but once registered, the FDA may conduct random inspections and can issue a Form 483 for non-compliance
- If the manufacturer is a foreigner, shall appoint an FDA US Agent representative as a local point of contact with the FDA
- List the device and register your company using FURLS system on the FDA website; pay fees for Establishment Registration and Listing, which must be renewed each year
- Now the manufacturer can sell the device in the US.
Process flow
Approval timelines
(MD/IVD) Time-to-market varies by regulatory submission and device type. The following timelines are based on Emergo’s experience, and actual timelines vary depending on the complexity of the regulatory submission and other FDA variables:
- Class I (510(k) exempt): 1 month
- Class II (510(k): 6 – 9 months
- Class III (PMA): 18 – 30 months
US FDA regulatory for similar medical devices
To mark a device as substantially equivalent, manufacturers need to compare their device with one or multiple existing devices and provide suitable facts to support the claim.
- The base device selected for the comparison is called “predicate”.
- Any available device which does not violate any of the FDA acts can be used as a predicate, including devices that recently cleared 510(k) notification.
A device is considered as SE in the following two scenarios while comparing it with the predicate:
- A device has the same proposed use with the same technological features as the predicate.
- A device has the same proposed use as the predicate with different technological features, but that does not raise different questions on the safety and effectiveness of the device, and the information submitted by a manufacturer is sufficient enough to validate that the device is at least as safe and effective as the existing legally marketed device.
Are you looking for regulatory support in the United States?
Credevo offers a wide range of drug/medical device development and regulatory services in the USA. Provide your requirement details below to connect with us and explore our services.
Reference
- https://www.fda.gov/industry/fda-user-fee-programs/medical-device-user-fee-amendments-mdufa
- https://www.einfochips.com/blog/an-overview-of-fda-regulations-for-medical-devices/
- https://www.regdesk.co/overview-medical-device-regulations-usa/
- https://www.pacificbridgemedical.com/regulatory-services/medical-device/product-registration/
- https://www.selectusa.gov/medical-technology-industry-united-states