Clinical Trial Requirements and Practical Scenario for Marketing Approval in China

Do you know any product segment where one does not get to encounter Chinese products? Well, hardly any. It has been the focus of the Chinese government for the past several decades to dominate various industrial sectors, including pharmaceuticals. While China has been the big production factory for generic drugs and API ingredients for many years, more and more discovery projects have now started going through China. In this article, let’s understand the regulations for drug development and conducting clinical trials in China.

The Chinese government has brought regulatory reforms recently that shake up the global pharmaceutical R&D sector immensely. Read our previous article on Chinese regulatory reforms about clinical research and pharmaceutical product registrations.
These regulatory reforms seem to create a friendly environment for pharmaceutical development and market access and might prove to catalysts for growth not just in China but worldwide. However, there are some uncertainties and challenges.
Top challenges with Chinese regulatory reforms for drug development & conducting clinical trials in China
The practical challenges with Chinese regulatory reforms for global pharmaceutical companies are
- Transparency, communication, and understanding the new regulatory system,
- Incorporating China in simultaneous global development,
- ICH guidelines integration,
- Adapting innovative standards for new technologies for innovative drug development,
- Drug life cycle management. For example. drug withdrawal system building which is encountered, with language and cultural differences issues.
Various factors like
- Concurrent use of both western and Chinese medicines,
- Pharmacogenetic variances,
- Lack of multidisciplinary team,
- Impact on disease management and
- Drug safety monitoring
are having an impact on drug development in China
In our earlier blog, ”Changing Clinical Trials Scenario In China” we talked about the changing regulatory policies in the last few years, with key highlights being the acceptance of data from overseas trials and reduction of the regulatory approval timelines.
But, practically, how this is evolving, will make a lot of difference.
Many pharma/CRO/regulatory consultants believe that it will take a while for the complete transformation to take place.
Recent interactions with experts on the matter tell a different story. Let’s talk about that in a minute after understanding the current changes and regulatory timelines.
The current clinical trial approval process in China
Let’s have a look at the best scenario and timelines associated with the IND application in China.
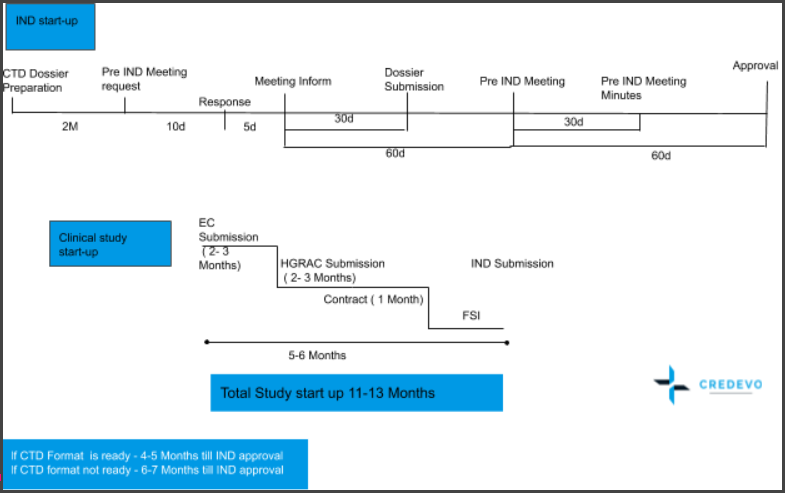
Key features of this timeline.
- IND review timelines are 60 working days, along with a simplified process.
- The process is simple.
- If the CDE doesn’t raise any query, then the IND application can be considered approved.
- In the best-case scenario, study start-up timelines are now 11-13 months for IND application, which includes dossier preparation, pre-IND meeting and application, EC and OHGRA approval, and contract finalization to enrolling the first patient in the trial.
Practical problems and questions
Now, coming back to the practical implications on the applicant’s experience, some significant steps, like
- Pre-IND meeting
- OHGRAC approval
can be a road-blocks, if not handled properly.
Many questions arise about a smooth process there.
- Are the timelines being followed? Has anyone got approval within these timelines?
- What is the data/information required for IND dossier submission?
- How much data needs to be given to regulators for pre IND meetings for the best meeting output?
- How to engage NMPA assessors during pre-IND to have a meaningful application?
- Along with the existing reforms, how to work without jeopardizing the speed and regulatory compliance of development projects?
Practical steps to have a successful Chinese clinical trial application process
In the recent DIA China 2019 event, many prominent professionals shared their experience with new regulations and NMPA because it was clear that there is a way for all applicants to have a smooth route through the application process.
1. The key is in the clarity!
Lack of clarity of these reforms and regulations can cost the delay in approval and increased R&D costs for companies. At the same time, a thoroughly defined clinical development strategy of their product is a must to face the regulators.
Are you planning to file a regulatory application for your product and unclear about regulations and processes to follow?
Get in touch with us today. Provide your preliminary details about the product application and your details in the below form to get started.
2. Communication, communication, and communication!
Communicating effectively with regulators at critical milestones can allow the sponsor to jointly solve difficult problems and issues which are not covered by technical guidelines.
Three types of Meeting requests were suggested by Administrative Provisions for Communication about Pharmaceutical Research & Development and Technical Review in June 2016.

Communication with CDE during the Pre-IND and Pre-NDA meetings are usually face-to-face meetings. The applicant can also have a chance to communicate via phone or email during the review process.
Problems faced in communications
Common problems faced while communicating with regulations are
- Preparation of information – how much data is sufficient?
- Provide sufficient background information to support the product in question, but not equivalent to IND submissions
- Provide sufficient information to support the issues discussed, including the original text of the document which is clear, hierarchical, and detailed
- Submit a conference communication slide Conducive to reviewers to improve efficiency.
- Way of asking a question – How to ask questions?
- The problem should be specific.
- There should be solid support for the proposed communication problem and can be judged by the management department.
- Avoid asking very broad or open questions.
- Communication process
- Applicants should have a process of self-analysis and evaluation of the proposed communication questions (not just the research results and the list of references).
- The applicant shall submit a preliminary answer to the questions raised and discussion based on science and practical operability.
- The applicant shall submit all meeting materials on time for meeting with the regulatory body. The regulatory body arranges meetings once they receive appropriate information to support the discussion and have sufficient time for review and discussion. Failing to do so may cancel the meeting application by the regulatory body.
- Communicating minutes of meeting
- The applicant shall communicate the minutes of the meeting with the management team.
Tips for effective communication
For improvising the quality of communication, regulatory authorities and sponsor need to continuously follow a functional set of rules applied in a timely and consistent manner, these are
- A mutual understanding of the terms and phrases used in communication: Use of them by both parties, the applicant, and the regulators shall be consistent.
- Legal or words to depict rules must be distinguishable from the words used for suggestions or nice-to-have issues.
- Due to differences in cultures, communication styles, preferences, and documentation needs, there is no “one line” of best communication methods, so continuous improvement is required.
- For time-critical matters, telephone communication between the sponsor and the FDA project manager may be relatively more effective than E-mail.
- The best practice is to follow up with the written communications after the telephonic conversation to document the decisions made during the contact period and agree on the action items.
Conclusion
While the recent reforms in the Chinese regulatory process for clinical trial application as well as product approval process appear overwhelming, there is a clear way possible to make it effective. It’s certainly not a straightforward or short-term process, but more of forming a habit of effective dealing with Chinese authorities. It’s important to gain experience for making submissions to NMPA and thus, achieve expertise; or hire an expert with experience and can deal with your projects.
It’s possible to have a successful and effective approval performed without compromising your project plans. The need is to be proactive and a great communicator.
Reference
- https://www.pharmaceuticalonline.com/doc/an-analysis-of-regulatory-reforms-in-china-s-pharmaceutical-market-0001
- https://cdn.ymaws.com/www.casss.org/resource/resmgr/wcbp_speaker_slides/2018_WCBP_LiMin.pdf
- https://cdn.ymaws.com/www.casss.org/resource/resmgr/cmc_japan/2018_CMCJP_MengYang.pdf
- https://www.pharmasug.org/proceedings/2019/SS/PharmaSUG-2019-SS-014.pdf
- https://www.lionbridge.com/blog/life-sciences/chinese-pharma-regulatory-changes-3-implications-for-the-world
- https://www.researchgate.net/publication/309689086_Challenges_for_drug_discovery_and_development_in_China
- https://www.researchgate.net/publication/315631213_Obstacles_and_opportunities_in_Chinese_pharmaceutical_innovation
2 thoughts on “Clinical Trial Requirements and Practical Scenario for Marketing Approval in China”
Comments are closed.