Preparing for Post-Patent Generic Launch: A Practical Guide to Bioequivalence Studies
When patents for Reference Listed Drugs (RLDs) expire, it opens the door for generic manufacturers to offer more affordable alternatives to brand-name drugs. However, successful entry into the market requires careful preparation ahead of the patent expiration. To demonstrate that a generic drug is equivalent to its branded counterpart, companies conduct thorough clinical research. This includes rigorous bioequivalence studies that compare how the generic formulation performs in the body relative to the original drug. This proactive approach not only helps in establishing credibility but also ensures that consumers have access to quality, cost-effective medication.

These studies, conducted by generic manufacturers and specialized contract research organizations, are known as bioequivalence studies. Bioequivalence studies examine food-drug interactions, formulation variations, and regulatory compliance to support generic approval. Manufacturers must prove their products match brand-name drugs in absorption rate and extent.
However, scientific expertise alone does not guarantee success in today’s competitive pharmaceutical landscape. Companies face a complex web of regulatory and legal challenges that can significantly impact their market entry strategies. To navigate these challenges successfully, manufacturers must first understand the fundamentals of the generic approval process. They must also know regulatory timelines that extend beyond patent expiration and execute strategic actions that ensure early market readiness.
This article focuses on post-patent entry after all patents and exclusivities expire, avoiding litigation complexities. We explore the essential steps: understanding bioequivalence foundations, navigating regulatory timelines, and implementing strategic actions for successful market entry.
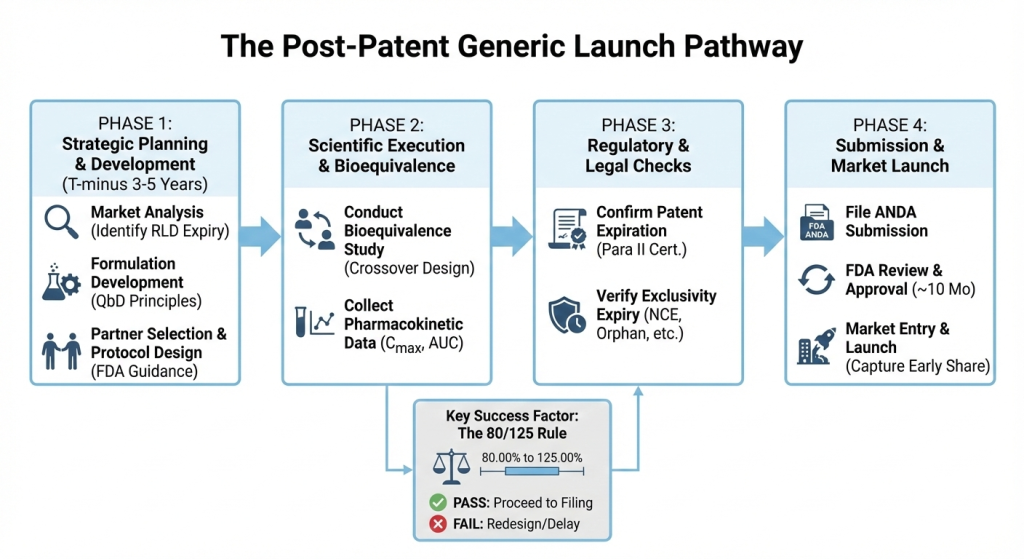
Understanding the foundations of bioequivalence studies and post-patent generic drug approval
When patents on reference-listed drugs (RLDs) expire, generic manufacturers follow a streamlined approval pathway focused primarily on demonstrating bioequivalence. This process emphasizes scientific validation and regulatory compliance without the legal complexities of patent challenges.
- Establishing bioequivalence through clinical studies
The first step is conducting bioequivalence (BE) studies, which demonstrate that a generic drug performs identically to its reference listed drug (RLD). These studies measure how quickly and completely the body absorbs the active ingredient and delivers it to the site of action (bioavailability). Researchers typically use crossover designs with healthy volunteers to compare how both drugs behave in the body.
Both the FDA and EMA accept randomized, crossover bioequivalence study designs for most immediate-release oral dosage forms. While study conduct principles remain consistent, region-specific guidance may influence subject numbers, fasting/fed conditions, and statistical considerations.
- Measuring critical pharmacokinetic parameters
During these studies, researchers collect specific pharmacokinetic (PK) data that form the basis of regulatory comparison as defined below:
- Cmax: Maximum plasma concentration achieved
- Tmax: Time to reach maximum concentration
- AUC0-t: Area under the curve from time zero to last measurable concentration
- AUC0-∞: Area under the curve extrapolated to infinity
Among these metrics, Cmax and AUC serve as the primary indicators for assessing bioequivalence, providing quantifiable evidence of how the drug behaves in the body. These measurements then become the foundation for demonstrating regulatory compliance.
These pharmacokinetic parameters are universally recognized across major regulatory agencies, including the FDA and EMA, as the primary metrics for assessing the rate and extent of absorption. Their consistent use supports international regulatory convergence in generic drug evaluation.
- Meeting the 80/125 regulatory standard
With pharmacokinetic data in hand, the next step is to demonstrate that the generic product meets the stringent regulatory standard known as the 80/125 rule. Under this standard, the 90% confidence interval for the ratio of geometric means (Test/Reference) must fall within 80% to 125% for Cmax, AUC0-t, AUC0-∞ . This range, which corresponds to a ±20% difference on a log-transformed scale, demonstrates that any difference between the generic and brand-name drug is not clinically significant. Once this scientific foundation is established, manufacturers can proceed to the regulatory submission phase.
- Filing the ANDA with Patent Certification
With bioequivalence data established, manufacturers file an ANDA under the Hatch-Waxman Act. They use Paragraph I (no patent filed) or Paragraph II (patent expired) certification for post-patent drugs.
This streamlined pathway allows generics to rely on the innovator’s safety and efficacy data:
Before filing an ANDA, it’s important to understand how this pathway differs fundamentally from the New Drug Application (NDA) process required for brand-name drugs, as detailed below:
| Aspect | NDA (New Drug Application) | ANDA (Abbreviated New Drug Application) |
| Purpose | Approval of new/innovator drugs | Approval of generic versions |
| Clinical Trials | Full Phase I, II, III trials required | Not required |
| Evidence Required | Safety & efficacy data | Bioequivalence to RLD |
| Review Time | ~10-12 months (standard) | ~10 months (goal) |
| Cost | $2-3 billion+ (including R&D) | $1-5 million |
| Exclusivity | Various (5-7 years for New Chemical Entity) | 180 days (first filer) |
In the European Union, regulatory authorities approve generic medicines through centralized, decentralized, or national procedures, depending on the product and market strategy. While terminology and submission routes differ from the ANDA pathway, the underlying requirement to demonstrate bioequivalence to the reference product remains consistent across regions.
This streamlined approach significantly reduces the time and cost required to bring generic medications to market, making generic development highly attractive from both a timeline and investment perspective.
- Regulatory Review and Approval: U.S. and Global Perspectives
Once a complete application is submitted with robust bioequivalence data, regulatory authorities review the submission to ensure compliance with quality, safety, and efficacy standards. In the United States, the FDA evaluates ANDAs with a goal review timeline of approximately 10 months. Similarly, the EMA and national European authorities assess generic applications with a focus on bioequivalence, manufacturing quality, and Good Clinical Practice compliance. While review timelines and procedural steps vary by region, scientific expectations remain closely aligned.
- Market entry and competition (add other regulatory)
After receiving FDA approval, generic manufacturers can immediately launch their products. In the post-patent environment, multiple generics typically enter simultaneously, creating robust competition that drives prices down significantly, often 80-85% below brand-name prices. This competitive dynamic benefits patients through improved medication affordability and access, fulfilling the core objective of generic drug policy.
Understanding regulatory exclusivity timelines
While the ANDA pathway focuses on scientific bioequivalence, generic manufacturers must also understand regulatory exclusivities that can delay market entry even after patents expire. These government-granted protections operate independently of patents and create mandatory waiting periods before generic competition can begin.
Understanding when these exclusivities expire is critical for timing bioequivalence studies and ANDA submissions. Starting development too early wastes resources if exclusivities block market entry. While starting too late causes manufacturers to miss the critical early launch window when prices are highest and market share is being established.
The regulatory exclusivity is independent of patents and varies by jurisdiction. The United States and the European Union have different frameworks. But the two systems can significantly delay the generic entry even after the expiry of the patent. This makes early regulatory information essential to development planning.
Types of Regulatory Exclusivities:
| Exclusivity Type | Duration | Region | Submission Restriction | Market Entry Restriction | Key Features |
| NCE (New Chemical Entity) | 5 years | United States | ANDA filing blocked for 4 years | Market entry blocked for 5 years | Applies to new active ingredients only. Exception: Paragraph IV filers can submit after 4 years if challenging patents. Cannot be shortened or challenged |
| Orphan Drug | 7 years | United States | ANDA filing blocked for 7 years | Market entry blocked for 7 years | Applies to drugs treating rare diseases (<200,000 patients). Blocks all competition, even if patents expire. Strongest form of exclusivity |
| Data Exclusivity | 8 years | European Union | Generic cannot reference clinical data for 8 years | Market entry blocked for 8 years | Part of EU’s “8+2+1” framework. Protects investment in clinical trials. Independent of patent status |
| Market Exclusivity | 10-11 years total | European Union | Generic can prepare application after 8 years | Market entry blocked for 10-11 years | Additional 2 years beyond data exclusivity. +1 year if new therapeutic indication approved. Allows ANDA preparation but not launch |
| Pediatric Exclusivity | +6 months | United States | Extends existing exclusivity by 6 months | Extends market protection by 6 months | Added to existing NCE or patent term. Granted for completing pediatric studies. Can be combined with other exclusivities |
For generic manufacturers, the key task is to find out when all patents and exclusivities will expire. Once manufacturers know the date/time, they must ensure their bioequivalence studies, ANDA preparation, and FDA review are completed on time. Understanding these regulations helps manufacturers plan their actions effectively.
Having established the scientific requirements and regulatory timeline considerations, we can now look at the specific strategic actions for successful market entry. These are discussed in detail below:
Key actions for generic manufacturers for post-patent generic entry
- Early planning is critical
To maximize market entry success, initiate formulation development and pilot studies well before patent expiration. Ideally, these studies can be started three to five years before the expected expiry date of the patent. Late entrants miss the critical early stage when prices are highest, and market share is being established. Starting early allows for a strategic positioning that can capitalize on this critical phase of high demand and profitability.
- Strategic CRO selection:
When considering potential Contract Research Organization (CRO) partners, it is essential to conduct a comprehensive assessment of their backgrounds. It is crucial to ensure that the CRO has a proven track record in the specific therapeutic areas relevant to your study. Partnering with the right CRO can significantly streamline project timelines and minimize risks. It ultimately aids in the successful execution of bioequivalence studies and facilitates a smoother pathway to market.
- Focus strictly on the 80/125 rule:
To ensure a smooth path to market entry, the 90% confidence interval for Cmax and AUC must remain strictly within the range of 80.00% to 125.00%. Adhering to this range helps prevent the need for expensive study repetitions. As it can cause delays of 6 to 12 months and compromise early launch opportunities.
- Utilize Product-Specific FDA guidance:
It is important to adhere to the FDA’s product-specific bioequivalence recommendations. As this ensures that your study design aligns with the most current regulatory expectations and effectively avoids common deficiencies that can result in Complete Response Letters. Following these guidelines can significantly enhance the quality and compliance of your submission.
Manufacturers often rely on product-specific, regulator-aligned study protocols to reduce design errors and avoid review delays.
To aid in reducing design errors and review delays, Credevo has curated 10 BE protocols for molecules that are approaching patent expiry. It can support effective study design and regulatory preparation for your studies.
Click here to learn more: https://credevo.com/s/bioequivalence-study-protocol-catalogue/
- Build quality through QbD principles:
Implementing Quality by Design (QbD) during formulation development and manufacturing processes is essential for creating robust and predictable outcomes. This approach helps ensure that products are consistently bioequivalent, thereby minimizing the risks associated with bioequivalence study failures and manufacturing deviations. By prioritizing QbD, organizations can enhance the reliability and quality of their products throughout the development lifecycle.
By executing these five key actions, generic manufacturers position themselves for successful post-patent market entry. This allows capturing significant market share while providing patients with affordable access to essential medications.
Conclusion
Post-patent generic markets offer substantial opportunities for manufacturers who execute with careful strategy and precision. By initiating thorough planning 3 to 5 years before the expiration of a patent, manufacturers can position themselves for success. This includes conducting comprehensive bioequivalence studies and implementing robust manufacturing systems to ensure product quality and regulatory compliance.
Are you looking for guidance in developing a bioequivalence protocol aligned with regulatory requirements?
Connect with our team to discuss your study requirements and related bioequivalence study considerations.
You can also explore our ready-to-use bioequivalence study protocols here:
Fill in the form below to learn more: