Japan – Clinical Trial Regulatory Process

There is an increase in interest for the drug developers to conduct clinical trials in Japan due to huge market size, clear regulatory guidelines, a crisp approval process, and other such factors. This calls for adequate awareness about the regulatory process required to start clinical trials in Japan.
Note: This information was last updated in February, 2023.

Japan is the world’s second large pharmaceutical market and accounts for approximately 10% of global drug sales and also the Japanese market is expanding year on year.
The world’s top pharmaceutical companies that operate in Japan enjoy annual growth rates ranging from 12% to 31%, in contrast with the expansion seen in other emerging markets.
This growth spurt attributes to the Japanese government’s multi-year focus to transform the drug approval process, regulations, and reimbursement.
Why choose Japan for your clinical trials?
There are several reasons why Japan is an attractive location for conducting clinical trials:
- Strong Regulatory Framework: Japan has a well-established regulatory framework for clinical trials, which provides clear guidelines for the development of new drugs and medical devices. The Pharmaceutical and Medical Devices Agency (PMDA) is the regulatory body responsible for the review and approval of clinical trials in Japan.
- Large Patient Population: Japan has a large population of around 126 million people, which provides a significant pool of potential study participants for clinical trials. Japan also has a high incidence of certain diseases such as gastric cancer and hepatitis C, making it an attractive location for clinical trials targeting these conditions.
- Highly Skilled Workforce: Japan has a highly skilled and experienced workforce, including doctors, researchers, and clinical trial coordinators. The country also has a strong tradition of research and development in the healthcare sector, with many world-class research institutions and pharmaceutical companies located in Japan.
- Access to Advanced Technologies: Japan is known for its advanced healthcare technologies, including imaging and diagnostic tools, which can enhance the efficiency and accuracy of clinical trials. These technologies can also help researchers to better understand disease mechanisms and patient responses to treatments.
- Government Support: The Japanese government has made efforts to promote clinical research in the country by providing funding and incentives for clinical trials. For example, the government offers tax incentives to companies conducting clinical trials in Japan, and has established programs to support international collaboration in clinical research.
Overall, Japan offers a favorable environment for conducting clinical trials, with a strong regulatory framework, large patient population, skilled workforce, advanced technologies, and government support.
Regulatory authority for clinical trials in Japan
The Pharmaceuticals and Medical Devices Agency (PMDA) is the regulatory authority under the Ministry of Health, Labour, and Welfare (MHLW).
The MHLW performs the following
- Conducts scientific review of applications for clinical trial and registration of drugs, medical devices, cosmetics, nutraceuticals (Health Foods), etc.,
- Monitors post-market safety of drugs and medical devices, and
- Provide relief compensation for sufferers from adverse drug reactions and infections caused by the use of drugs or biological products.
Clinical Trial Approvals
- Pharmaceutical Affairs Law (PAL) governs the clinical trials in Japan.
- In accordance with Article 15 of the Good Clinical Practices, foreign sponsors should appoint an in-country representative.
- The approval process for clinical trials is sequential.
Human Ethics Committee in Japan
- In Japan, it usually takes four to eight weeks to obtain Institutional Review Board (IRB) approval.
- Japan also operates a local IRB process and has a Japan Good Clinical Practice (J – GCP) guideline.
- J- GCP is harmonized with the ICH – GCP guideline, though there are some considerations specific to Japan – GCP.
- As per J-GCP requirements, the site director needs to ask IRB whether the clinical trial is feasible to perform.
- Each site should use its IRB, but in some cases it is also possible to get approval from external IRBs.
- The head of each trial site should take responsibility to sign any contract and oversee or monitor the clinical trial.
Are you looking for regulatory support in various stages of drug development? then, write to us at inquiry@credevo.com or provide your query details in the form below to connect with us.
Clinical Trial Agreements (CTAs) for clinical trials in Japan
There is no specific requirement to obtain the approval of a CTA. However, the parties are required to enter into a
CTA providing for the matters prescribed in Article 13 of the GCPs.
Which include
- Date of the agreement
- Name and address of the sponsor
- If responsibilities relating to the clinical trial are to be outsourced to a contract research organization (CRO), name and address of the CRO and the scope of outsourcing
- Name and address of the medical institution carrying out the clinical trial, the persons responsible for the agreement and position of the investigator
- Term of the clinical trial
- Matters concerning the management of the tested product, maintenance of records, notifications, the maintenance of confidentiality regarding trial subjects and the costs of the clinical trial.
- Provision to the effect that the medical institution will carry out the clinical trial in accordance with the clinical trial plan
- Matters concerning indemnity for health hazard to trial subjects
- Other necessary matters
The sponsor must take necessary measures like arranging insurance coverage beforehand, provide compensation to trial subjects (including those arising from the operations of a CRO) caused by the clinical trial (Article 14 of the GCPs).
Clinical Trial Notification (CTN) for clinical trials in Japan
- Clinical trials sponsors must follow the standards specified in ordinance passed by the Ministry of Health, Labour and Welfare (MHLW).
- Sponsors of clinical trials must submit the clinical trial protocol to the Pharmaceuticals and Medical Devices Agency (PMDA).
- The persons who submits the protocols shall not sponsor the clinical trial after 30 days from the date of submission and in such case, the minister will review the clinical trial protocol to prevent jeopardizing public health and hygiene.
Drugs that come under Clinical Trial Notification (CTN)
Here are some categories of drugs that come under the Clinical Trial Notification (CTN) scheme.
- The drugs with active ingredients and but differ from approved drugs.
- Drugs with the same active ingredients, but the route of administration differs from the approved drugs.
- The drugs that differ from combination ratio of active ingredients, or indications or dosage and administration of approved drugs.
- Medicines with active ingredients that are the same ingredients of drugs approved as a new molecular entity, but for which the re-examination period is still not expired.
- For biologics.
- The drugs manufactured using recombinant technology.
Documents for Clinical Trial Notification (CTN)
Here is the list of documents required for Clinical Trial Notification (CTN).
- A statement that scientifically justifies the reason for sponsor to intiate the clinical trial.
- A clinical trial protocol.
- Informed consent form.
- A Case Report Form (CRF) sample.
- The current Investigator’s Brochure (IB).
Note: All the documents need to be in the Japanese language.
Timeline for CTN
- The sponsor shall submit the first CTN of trial drugs at least 31 days before the day of planning a contract with the medical institutions.
- The applicant shall submit other notifications other than those specified above, at least two weeks before the day of the planning contract with the medical institutions.
The clinical trial approval process in Japan
- The clinical trials approval process is sequential in Japan, where the sponsor first needs to obtain IRB approval before making an application for clinical trial approval.
- The sponsor shall submit an initial notification and a study protocol to the PMDA before they enter any contract with any institution or site to conduct a clinical trial.
- The submission requirements for clinical trials are similar to those of other countries such as clinical trial protocol, informed consent form, names of all investigators, ethical approvals, etc.
- The PMDA assesses all these documents to check whether the proposed study complies with the regulatory requirements. In this review, the PMDA considers the science, proposed data collection, analysis techniques, patient safety, and ethics.
- After assessment, the PMDA then implements the “Clinical trial consultation system” where it gives guidance and advice related to study protocols.
- Clients may choose whether or not to request a consultation before they submit a CTN.
- However, the PMDA strongly encourages sponsors to utilize the consultation system, as it may reduces the review time for application.
- Clients who plan multinational trials with some sites in Japan may choose to skip the pre-CTN consultation.
- However, they can request a session before they file the NDA to confirm that their trial data is adequate.
- The number of consultations required will also vary by therapeutic category.
- The time for the consultation request is approximately two months.
- The PMDA ensures to assing the same reviewers to a study throught the lifecycle. As it eliminates the need to acquaint a new reviewer with the clinical study.
- Clinical trial approval is default. Which means, if the client does not receive any queries from PMDA in the specified period, the sponsor can commence the trial.
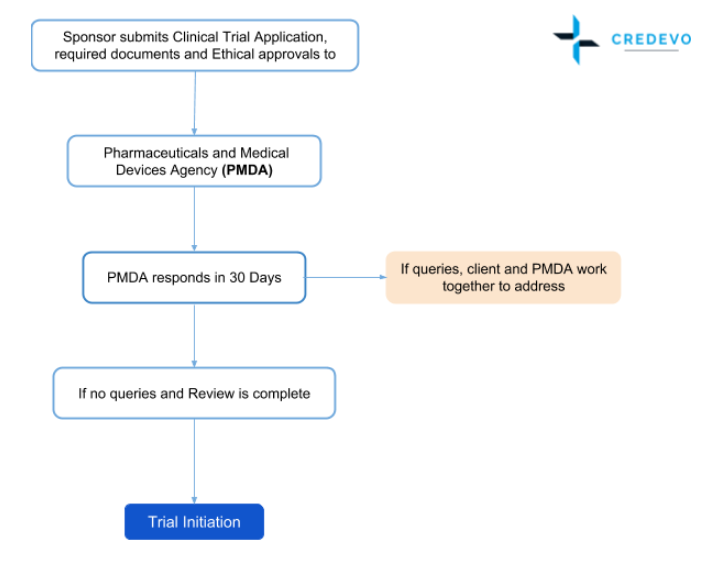
Timelines for clinical trial approval
- The consultation with the PMDA takes about three months.
- The sponsor shall submit the notifications for drugs with new active ingredients or new routes of administration, or new combination drugs at least 31 days before the start date of the trial.
- For some other categories of drugs other than those mentioned above, the sponsor needs to submit application at least two weeks before the planned date of the trial.
In-country caretaker for clinical trials in Japan
A sponsor initiates, operates, monitors, prepares SOPs, prepares clinical trial protocol, completes studies on the quality, and manages a clinical trial. However, if the sponsor is not located in Japan, can appoint an in-country caretaker who resides in Japan.
In-country caretaker
An in-country caretaker can carry out the necessary procedures for sponsoring a clinical trial.
Functions of an in-country caretaker
- Plan development strategy
- Conduct Phases I to III trials
- Prepares approval application form, approval application summary and attach documents as well as application
- Post-market usage result survey
- Carry out the responsibilities of the in-country clinical caretaker and designated marketing approval holder (D-MAH)
- Trial-related services other than those mentioned above
- Consultation regarding development, approval/license and statistical analysis
- Quality assurance/audit and consultation regarding quality assurance/audit and training services
- Cooperation for the establishment of a clinical trial system.
However, the sponsor retains ultimate legal responsibilities.
Are you looking for an in-country caretaker who handles all your clinical trial responsibilities?
Complete the form below to connect with us and discuss your clinical trial needs in Japan.
What is “Harmonize by doing”?
- It is a collaboration programme between the regulators US FDA and Japan PMDA.
- The collaboration between the Japanese and United States regulators (PMDA and FDA) aims to harmonize regulatory requirements and guidelines for the development and approval of new drugs and medical devices.
- This approach can accelerate the development and approval of new products while maintaining ethical and regulatory standards.
- Initiatives such as the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) promote the development of common technical standards and guidelines for clinical trials and product assessment worldwide.
How does “Harmonize by doing” benefit the sponsors?
Harmonization of regulatory requirements and guidelines between countries can benefit sponsors by reducing the
- regulatory burden,
- improving the quality and consistency of clinical data, and
- providing a more efficient and streamlined regulatory framework.
This can lead to cost savings, faster timelines, and a more predictable approval process for new drugs and medical devices.
Top 4 challenges to conducting clinical trials in Japan
While Japan’s clinical trial infrastructure is rapidly growing, still there are some unique challenges for sponsors to overcome.
Japan – Good Clinical Practice (J-GCP) J-GCP is harmonized with the international standard (ICH-GCP) and governs the standards for Good Clinical Practice for clinical trials in Japan. But, the J-GCP is more comprehensive compared with the ICH and includes additional requirements that often slow the development process.
Language barrier: Though most Japanese physicians can read and understand the clinical trial-related documents written in English, other site-level personnel e.g., clinical research coordinators, pharmacists, and administrative assistants may not have the same level of language fluency.
Limited availability of investigators: With an increase in health care expenditure, and a rapidly aging population, the Japanese government was forced to limit the spending of funds on health care. As a result, there is short of clinicians at all levels, and out of them, only a few physicians have time to devote to clinical research.
Limited incentives for investigators & patients: Unfortunately, the limited availability of Japanese investigators is exacerbated by the lack of incentives for participation. Since every Japanese patient has full medical coverage, enrolment in clinical trials does not provide a means to access a higher level of care.
Credevo can help you to conduct clinical trials in Japan. Contact us at inquiry@credevo.com or fill out the form below to get regulatory support.
Still have some questions?
Japan offers a lot to all kinds of drug / medical device / biotech companies in terms of market expansion. It’s always interesting to consider Japan in your global strategy.
Having reviewed the regulatory process and considering benefits / challenges in Japan, if you have more questions, feel free to ask us.
One thought on “Japan – Clinical Trial Regulatory Process”
Comments are closed.