India is ready for your clinical trials. Are you?

A country with a large pool of patients, highly skilled Investigators, and supportive regulations to promote clinical research is a desired destination for any organization for clinical trials. India has a lot more to offer besides these advantages, enabling you to enjoy the timely completion of clinical trials and cost benefits. (India offers a 35 to 60 percent cost advantage compared to the Western markets.)
This article was last updated in May 2023.

Top 5 Advantages of conducting clinical trials in India
- A large pool of patients,
- Highly skilled Investigators,
- Supportive regulations to promote clinical research,
- Timely completion of clinical trials, and
- Cost benefits
Despite its good standing in clinical research, India needs to utilize a significant part of its potential to conduct clinical trials.
- According to a report by the Indian Council of Medical Research (ICMR), the number of registered clinical trials in India increased from 141 in 2005 to a peak of 539 in 2010, before declining to 183 in 2018.
- However, despite the decline in the number of trials, India still ranks among the top 10 countries in terms of the number of registered clinical trials.
- The majority of trials conducted in India are sponsored by multinational pharmaceutical companies, and the therapeutic areas that have the highest number of trials are oncology, cardiology, and endocrinology.
There was a time when Indian regulations were bringing drastic changes with concerns for those wishing to conduct clinical trials. Not anymore. With rules clarified and processes smooth-lined, India has prepared itself for the next phase of clinical research growth.
Let’s have a look at current regulatory requirements for conducting clinical trials in India.
Some salient points.
- Clinical trial application language is English
- Regulatory and Ethical approval is parallel
- In-country sponsor/representation is required
- Clinical trial registration is required
Regulatory authority for clinical trials in India
The Central Drugs Standard Control Organization (CDSCO) is the National Regulatory Authority in India. Its equivalent counterparts elsewhere include
- The United States Food and Drug Administration (US FDA),
- Health Canada (HC) and the
- European Medicines Agency (EMA).
The Drugs Controller General of India (DCGI), an official of the CDSCO, is the final regulatory authority for the approval of clinical trials in the country.
DCGI office is also responsible for inspections of trial sites, sponsors of clinical research, and manufacturing facilities in the country.
In case of noncompliance, the DCGI may reject, discontinue, or cancel the clinical trial permission or debar the investigator(s), sponsor including, his representative, to conduct any clinical trial in the future.
The DCGI conducts a review and approval process in parallel with the EC review, except in clinical trials for academic/research purposes that are non-regulatory.
DCGI approval fee
Requisite fees as per the provisions of the Drugs & Cosmetics Act and Rules are
- For feasibility study (i.e. Safety and efficacy study); which is equivalent to Phase I trials in case of drugs: INR 50, 000/- (approx 760 USD)
- For the pivotal study (i.e. Confirmatory trials); which is equivalent to Phase II/III trials in case of drugs: INR 25, 000/- (approx 380 USD)
How & when to start a clinical trial in India?
Prerequisites to conduct a clinical trial in India
- Permission from the Drugs Controller General, India (DCGI).
- Approval from the respective Ethics Committee where the sponsor plans the study.
- Register with the Indian Council of Medical Research (ICMR) ICMR maintained CTRI website
The sponsor must submit the clinical trial application to both the DCGI and the EC. The EC must grant a separate approval for each trial site to be used, and the sponsor must DCGI for each approval.
The investigator must ensure to conduct clinical trials as per the rules outlined below
- In compliance with an EC and a DCGI-approved protocol
- In the case of IISs with ‘new drugs’, DCGI approval is no longer needed; only an EC approval is required
- In compliance with GCP guidelines
- All applicable regulations.
Ethics committees
- India has a decentralized process for the ethical review of clinical trial applications and requires ethics committee (EC) approval for each trial site. The majority of ECs are at clinical or academic institutions and hospitals.
- The Investigators and administrators of the academic institutes shall register their Institutional Ethics Committees (IECs) with the central licensing authority and renew the registration at the end of 3 years.
Institutional Ethics Committee approval
- All clinical trials require IEC approval.
- A recent regulatory change concerning IISs is that academician who carry out clinical trials with new drugs no longer require approval from the DCGI to conduct the clinical trial. The IEC approval is sufficient.
- If the IEC feels that there could be a potential overlap between the academic and regulatory purposes of the clinical trial, they should notify the office of the DCGI.
- If the IEC does not hear any response from the DCGI within 30 days, then the IEC can be presumed that no permission is necessary from the licensing authority.
Projects submitted essentially undergo two broad types of review
- Complete board or full committee review – for all projects that present more than minimal risk or
- Expedited review for projects that pose minimal risk. e.g., leftover clinical samples.
Projects like systematic reviews or meta-analyses may also be exempt from review.
IEC/IRB review process
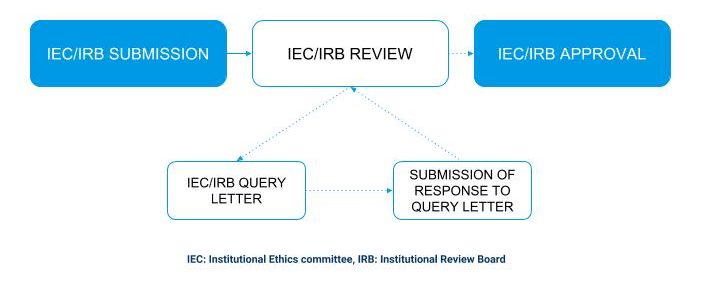
Clinical trial registry – India (CTRI)
Yes, The Clinical Trials Registry- India (CTRI) is a free online public record system to register clinical trials in India (www.ctri.nic.in).
Clinical trial registration in the CTRI is made mandatory by the Drugs Controller General (DCGI).
Clinical trial registration involves
- Public declaration and identification of trial investigators,
- Sponsors,
- Interventions,
- Patient population etc., before the enrollment of the first patient.
- Submission of the ethics committee and DCGI approval (if applicable) is essential to register in the CTRI.
If the trial is already registered with another primary register then why need to register again with the CTRI?
The sponsor needs to register the clinical trial in CTRI, to perform clinical trials in India. The CTRI captures data specific to the Indian arm of a clinical trial, for example, site and PI details, the ethics committee name and approval status, the target sample size in India, start date in India, etc. However, Registrants must quote the Registration number of the other Primary Registry number in the Secondary ID section of the trial registration data set.
Multi-regional trials
The sponsor shall register the multi-regional clinical trials with CTRI for the internationally registered trials, where India is one of the centers.
In the CTRI, the following details are necessary.
- Indian investigators,
- Trial sites,
- Indian target sample size and
- Date of enrollment
The fee to register clinical trials or access/view registered trials
There is no charge to register for a trial. Registered trials are also freely accessible to the public.
When is a trial considered to be registered?
A trial is registered when an internationally agreed set of information about the design, conduct, and administration of clinical trials is publicly available in a Primary Registry before the enrollment of the first patient
What is the UTN?
- The Universal Trial Number (UTN) (earlier known as the UTRN) is a unique number that aims to facilitate the unambiguous identification of clinical trials registered in WHO Primary Registries and displayed on the WHO International Clinical Trials Registry Platform (ICTRP) Search Portal. It is not a registration number.
- The sponsor shall obtain the UTN early and shall permanently attach it to the clinical trial.
What are other expectations and requirements?
Clinical trial site
The sponsor is responsible to select the investigator and institution for the clinical trial, taking into account the appropriateness and availability of the study site and facilities.
Informed consent from participants
Investigators must ensure that
- The sponsor shall obtain informed consent from all participants in a clinical trial, and
- Audiovisual recording of the informed consent process for clinical trials that involve vulnerable participants (children or mentally challenged patients, for example) and a new chemical entity or a new molecular entity.
Submission and approval process
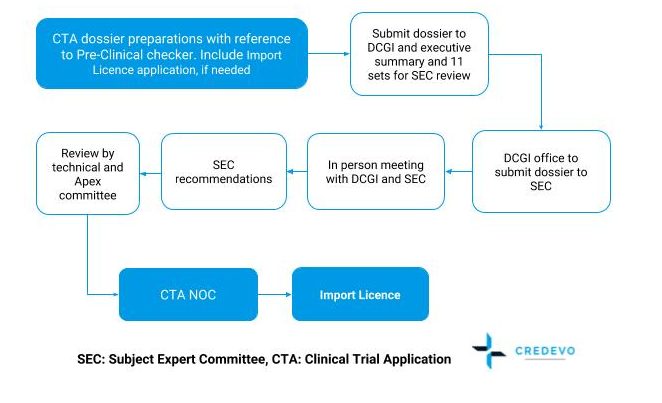
SEC: Subject Expert Committee, CTA: Clinical Trial Application
Timeline for approval
The approval timeline is approximately 4-5 months.
Clinical trials of medical devices
- In India import, manufacturing, sale, and distribution of Medical devices are regulated under the Drugs and Cosmetics Act and Rules.
- The Indian Council of Medical Research (ICMR) is the apex body that is responsible for the formulation, coordination, and promotion of biomedical research.
What are the requirements to conduct medical device clinical trials?
Global Regulatory status of the device: (specifically in 5 GHTF countries- USA, Australia, Japan, Canada, and European Union)
- Clinical trial in each participating country
- The regulatory status of a device in other countries
Ethics committee approval letters (if available): Information on the Ethics Committee constitution and format for approval of Ethics.
Technical data to submit
- Design analysis data.
- Biocompatibility data.
- Protocol for carrying out biocompatibility study
- Tests to establish biocompatibility (along with test reports) and rationale to select those tests
- Summary report of the biocompatibility study including the conclusion of the study.
After registration of trials
After a trial registration, triallists need to regularly update the trial status or other aspects, as the case may be. All updates and changes will be available for public display.
Import/Export of biological samples
- The requirement of NOC for the export of biological samples is no longer applicable.
- Customs authorities at the port of entry/exit shall permit the export of biological samples without prior approvals from any government agency, provided the concerned firm submits an undertaking confirming adherence to rules for the safe transfer and disposal of biological samples.
- That is a positive step to facilitate ease to conduct clinical research in the country and has further simplified the administrative process for study conduct.
Do you need support for conducting clinical trials in India?
Do you have any questions or do you need support in conducting clinical trials in India? Please provide your requirements details below so that we can connect with you and explore our services.